Article Text

Abstract
Holoprosencephaly (HPE) is a clinically variable and genetically heterogeneous central nervous system (CNS) malformation. Alobar HPE, which is its most severe form, is associated with a poor prognosis. At the milder end of the HPE spectrum microcephaly, hypotelorism, and single central maxillary incisor may be recognised. Currently, four genes have been identified for this condition. These include Sonic Hedgehog (SHH) on chromosome 7q36, which is thought to be responsible for a significant proportion of autosomal dominant HPE. We report an index case with alobar holoprosencephaly caused by an SHH mutation and six members of his family over two generations with this mutation, with a broad range of clinical presentation, including attention deficit hyperactivity disorder (ADHD). The combination of microcephaly, hypotelorism, subtle midline facial anomalies, and ADHD within a sibship should alert the physician to the possible diagnosis of HPE.
- holoprosencephaly, attention deficit hyperactivity disorder
- sonic hedgehog
- gene
- ADHD, attention deficit hyperactivity disorder
- CNS, central nervous system
- CT, computed tomography
- HPE, holoprosencephaly
- MRI, magnetic resonance imaging
- Ptch, patched transmembrane receptor
- Smo, smoothened transmembrane receptor
Statistics from Altmetric.com
- ADHD, attention deficit hyperactivity disorder
- CNS, central nervous system
- CT, computed tomography
- HPE, holoprosencephaly
- MRI, magnetic resonance imaging
- Ptch, patched transmembrane receptor
- Smo, smoothened transmembrane receptor
Holoprosencephaly (HPE) is a clinically and genetically heterogeneous malformation of forebrain development.1 The most severe form is called alobar HPE, where there is failure of division of the telencephalon into two cerebral hemispheres with a single ventricle.2 These children can present with severe craniofacial anomalies such as cyclopia, or premaxillary agenesis.3 Less severe forms of HPE, such as semilobar or lobar HPE can present with mild facial dysmorphism such as hypotelorism, iris coloboma, absent or abnormal upper labial frenulum, single maxillary central incisor, or cleft palate.4,5 Some patients with these forms of HPE may have no obvious craniofacial anomalies.4
Most cases of HPE are sporadic but familial forms have been described. These usually show autosomal dominant inheritance with reduced penetrance and variable expression.1,6 At the present time 12 loci for HPE have been identified and genes at four loci identified.7,8 These include Sonic Hedgehog (SHH) gene at 7q36, ZIC2 at 13q32, SIX3 at 2p21, and TGIF at 18p11.3.9–12 Mutations in these genes have been identified in sporadic and autosomal dominant forms of HPE.8–12
Mutations in SHH account for a significant proportion of autosomal dominant HPE.13 Although sporadic forms of HPE are more frequent than familial forms, SHH mutations have been identified more frequently in familial (autosomal dominant) HPE than sporadic HPE.8SHH is a homologue of the Drosophila hedgehog (hh) gene, which is a segment polarity gene.14 Studies in mice have shown that shh is expressed in a large number of tissues including notochord, ventrolateral midbrain, ventral forebrain, gut endoderm, branchial arches, posterior distal limb mesenchyme, testis, and penis.14 In Drosophila the hh protein is the ligand for a transmembrane receptor called patched (Ptch).15 In the absence of hh protein Ptch inhibits another transmembrane receptor called smoothened (Smo). When hh binds to Ptch, Smo is released from inhibition, which activates other intracellular signalling pathways.16 This hh/patched signalling pathway is conserved from Drosophila to mice.17 In humans the downstream target genes for SHH include the GLI factors, and the WNT and BMP gene families.18
We describe a family in which several members over two generations were found to have a missense mutation in SHH with remarkable variability of expression.
CASE REPORT
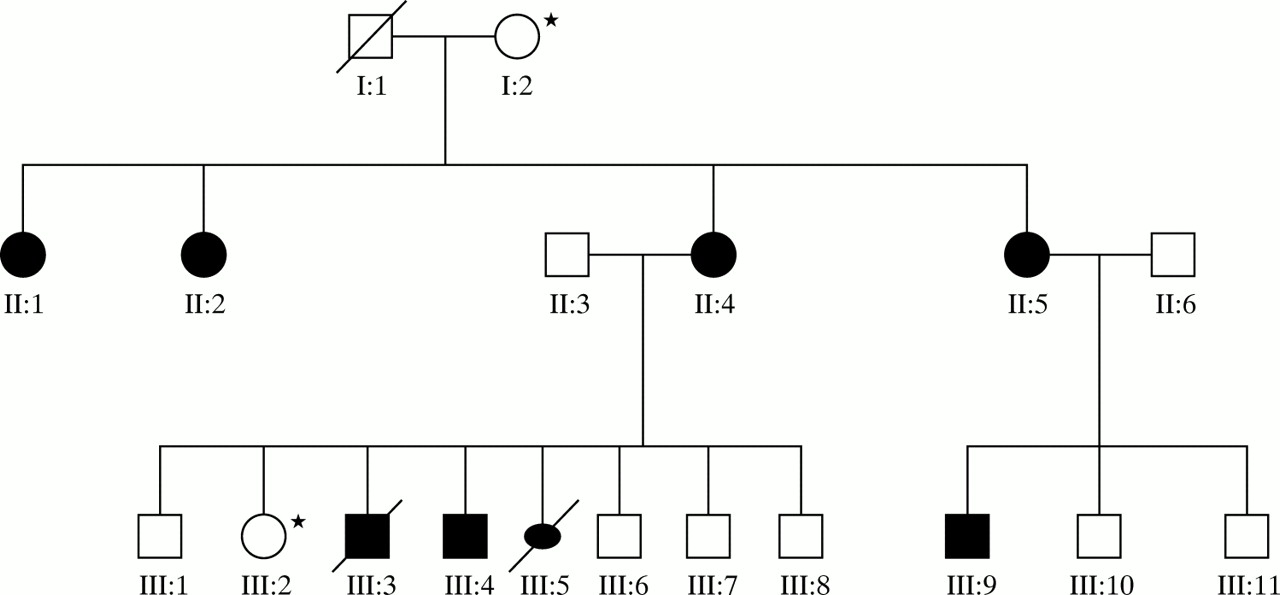
Figure 1 shows the pedigree of this family. The family was ascertained following the identification of HPE in the index case (III:3) by antenatal ultrasound.

Family tree depicting three generations with inherited SHH mutation. Starred family members were tested but were negtive for SHH mutation.
Case 1 (III:3)
This fetus was the product of the third pregnancy of non-consanguinous white parents. A detailed fetal anomaly scan at 20 weeks gestation had shown alobar HPE. The pregnancy was terminated at 21 weeks gestation and postmortem examination showed a male baby with microcephaly, hypotelorism, premaxillary agenesis, and alobar HPE (fig 2). Fetal karyotype was normal. A novel missense mutation in SHH was identified in fetal DNA. This mutation had resulted in a substitution of thymine for an adenine residue at nucleotide position 263 of SHH, resulting in the substitution of the amino acid aspartic acid for valine at position 88 of the SHH peptide.13

The index case (III:3), showing the midline cleft and hypotelorism.
Case 2 (III:4)
This is an older sibling of case 1. He was born at term with a birth weight of 2.94 kg (9–25th centile) and a head circumference of 34 cm (9–25th centile). He had a right sided cleft lip, cleft palate, inferior iris coloboma, sensorineural hearing loss, and single palmar creases. He had feeding difficulties with failure to thrive, global developmental delay, and postnatal development of microcephaly with his head circumference running parallel to but 2–3 cm below the 0.4th centile. At 3 years of age he was assessed using the Schedule of Growing Skills II.19 This showed that he was functioning at about the 18–24 months level in all areas except visual skills (wearing glasses) where he was functioning at an age appropriate level. When this assessment was repeated at the age of 36 months his locomotor, manipulative, speech and language, and interactive/social skills were at the 24 months level. His visual skills were at the 48 months level, and his hearing and language and self care/social skills were only at the 18 months level. A cranial magnetic resonance imaging (MRI) scan at this time was normal.
At the age of 6 years and 3 months he is hypotonic and clumsy, with mild learning difficulties. He also has problems with polydipsia and polyuria and drinks at least four litres of fluid a day. He has significant problems with enuresis but his early morning plasma and urine electrolytes and osmolality are normal. He has a clinical diagnosis of attention deficit disorder that has responded well to low dose methylphenidate (10 mg morning, 5 mg midday) with improvement in his functioning at school. Assessment using the NEPSY20 showed an attention executive score on the 21st centile, sensorimotor score on the 2–10th centile, and memory score on the 2nd centile. Genetic testing showed that he also had the Asp88Val SHH mutation that had been identified in his sibling with alobar HPE (figure 3).

Case III:4.
Case 3 (III:9)
This child is the maternal first cousin of cases 1 and 2. He was born at 34 weeks gestation by emergency caesarean section. His birth weight was 2.2 kg (50th centile) and his head circumference 28 cm (0.4th centile). He was noted to have hypospadias at birth. Postnatally his head circumference fell below the 0.4th centile and continued to grow 2–3 cm below the 0.4th centile. His height grew along the 0.4th centile and his weight just below the 0.4th centile. At 9 months his development was thought to be normal but at the age of 3 years concerns were raised about his development, particularly with regard to language. He was found to have notable hypotelorism. A Griffith21 assessment at 3 years and 9 months showed delays, particularly in speech and language where he performed at less than a 2 year level. He performed at a 2–2.5 year level for all other areas.
The Griffith assessment was repeated at the age of 45 months. His gross motor skills were at the 39 month level, personal social skills at 27 months, speech/language at 22 months, eye–hand coordination at 28 months, performance at 40 months, and practical reasoning at the 26 months level. His understanding and level of attention limited the assessment. He was unable to complete an NEPSY20 assessment as he had an attention/executive score of <1st centile, with very poor auditory attention and memory skills. His main difficulties were around hyperactivity, impulsive behaviour, and poor concentration, which impaired his performance in the classroom. He responded well to a trial of low dose methylphenidate (10 mg morning, 5 mg midday) with significant improvement in his abilities to follow instructions and complete tasks at school. He did not have a cranial MRI scan but genetic testing confirmed that he too had the Asp88Val SHH mutation. (figure 4)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Case III:9 and his mother (II:5). Another child in this family also has microcephaly, hypotelorism, and similar attention difficulties but has not been tested.
Case 4 (II:4)
The mother of cases 1 and 2 had learning difficulties as a child and also had problems with concentration. On examination, her head circumference was 2 cm below the 0.4th centile. She had mild hypotelorism and a high arched palate. She was also shown to have the Asp88Val SHH mutation (figure 4).
Case 5 (II:5)
The mother of case 3 had a history of mild learning difficulties and received some extra support at school. On examination her head circumference was 51 cm (<0.4th centile) and her height was 149 cm (0.4–2nd centile). She had mild hypotelorism and a high arched palate. She too tested “positive” for the Asp88Val SHH mutation (figure 4).
Case 6 (II:2)
This was the younger sister of cases 4 and 5. She was being treated for systemic lupus erythematosis. Her head circumference was on the 3rd centile and her height was on the 50th centile. She was also found to have inherited the familial SHH mutation like her two older sisters.
Case 7 (II:1)
This was the youngest sister of cases 4, 5 and 6. She had a history of moderate learning difficulties requiring education at a special school and lived with her mother. She had long-standing torsion dystonia, right facial hemiatrophy and a stable scoliosis. She also had in toeing of her right leg with sensory disturbance. Her cranial computed tomography (CT) scan was normal. She was thought to have an autosomal dominant form of dystonia as she had three maternal uncles who also had torsion dystonia. She tested positive for the Asp88Val SHH mutation.
The children's maternal grandmother (I:2) was also tested but shown not to carry the familial SHH mutation. It was therefore concluded that the mutation had been inherited from the maternal grandfather (I:1), who had died in his forties. No further information was available about this man.
DISCUSSION
This family highlights the intrafamilial variability of expression of an identical missense mutation in SHH (Asp88Val). The amino acid residue aspartic acid is located in the N-terminal signalling domain (SHH-N) at an invariant position in the hedgehog family of proteins and conserved from human, mouse, chicken, zebrafish SHH to Drosophila hedgehog. Although the functional effects of the Asp88Val change have not yet been studied, this mutation presumably leads to alterations in biological activity of SHH-N.8 The phenotypic variability in this family has been described in smaller kindreds previously, but to our knowledge this is one of the largest kindreds described. In this family the same SHH mutation was associated with alobar HPE in the index case but HPE “microforms” in other family members. Craniofacial malformations that are seen in individuals with normal neuroimaging, who are at risk of having children with HPE are called HPE “microforms”. These include microcephaly, ocular hypotelorism, iris coloboma, mid face hypoplasia, congenital nasal pyriform aperture stenosis, absent or abnormal upper labial frenulum, and single central maxillary incisors. In case 2 the ocular hypotelorism was an HPE microform as he had normal brain imaging and was shown to have inherited the familial SHH mutation. Case 3 also had hypotelorism but cranial CT or MRI scans were not performed and he could have had a mild form of HPE (semilobar or lobar). The microcephaly that was seen in cases 3, 4, and 5 was likely to have represented an HPE microform, but CNS imaging was not performed in these cases. Developmental delay and learning difficulties can also be considered microforms of HPE, as they can be seen in individuals with normal neuroimaging, who are at risk of having affected children with HPE (cases 2–5 and 7). In this family the phenotype ranges from very mild microcephaly and no clinical symptoms to an infant with alobar holoprosencephaly. Both children with the SHH mutation had attention difficulties. These have not previously been described as a microform of HPE, but may in fact just be a delay in maturation as both children improved significantly with age.
Non-penetrance has also been described in autosomal dominant forms of HPE. Obligate mutation carriers in such families may be asymptomatic with no craniofacial anomalies. This has been confirmed in families with autosomal dominant HPE with SHH and SIX3 mutations. Given the intrafamilial clinical variability in kindreds carrying an SHH mutation, we speculate that the other gene acting in the same or different developmental pathways might act as modifier for expression of the HPE spectrum. Interestingly the HPE patients with an SHH mutation were also identified with an alteration in a second gene which acts on brain development.13 Because of the non-penetrance in some HPE families, genetic counselling can be extremely difficult. Autosomal dominant HPE is estimated to have a penetrance of 70%.22 This means that the risk of anatomical HPE or microforms of HPE in the offspring of an obligate carrier of autosomal dominant HPE is about 35% (16–21% would be expected to have a severe form of HPE and around 13–14% would have mild HPE or microforms of HPE). Those with severe forms such as alobar or semilobar holoprosencephaly may be identified antenatally by fetal ultrasound scanning, but those with mild HPE or HPE microforms cannot be identified in this manner. These children, as shown by the manifestations in this family, may have a number of significant clinical problems and yet normal cranial morphology. These problems may include developmental delay, learning difficulties, cleft lip and palate, and microcephaly. Following the birth of a child with HPE, the parents need to be examined carefully for the microforms of HPE in order to provide them with an accurate recurrence risk for HPE or HPE microforms in another pregnancy. Although mutation analysis is not routinely available for autosomal dominant forms of HPE at present, this family illustrates the importance of combined clinical and molecular assessment to facilitate correct diagnosis and the provision of appropriate genetic counselling.