Revisiting the role of acetylcholinesterase in Alzheimer’s disease: cross-talk with P-tau and β-amyloid
- 1 Instituto de Neurociencias de Alicante, Universidad Miguel Hernández-CSIC, Alacant, Spain
- 2 Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas, Madrid, Spain
- 3 Menzies Research Institute, University of Tasmania, Hobart, TAS, Australia
- 4 Centro de Biología Molecular “Severo Ochoa,” Universidad Autónoma de Madrid-CSIC, Madrid, Spain
A common feature in the Alzheimer’s disease (AD) brain is the presence of acetylcholinesterase (AChE) which is commonly associated with β-amyloid plaques and neurofibrillary tangles (NFT). Although our understanding of the relationship between AChE and the pathological features of AD is incomplete, increasing evidence suggests that both β-amyloid protein (Aβ) and abnormally hyperphosphorylated tau (P-tau) can influence AChE expression. We also review recent findings which suggest the possible role of AChE in the development of a vicious cycle of Aβ and P-tau dysregulation and discuss the limited and temporary effect of therapeutic intervention with AChE inhibitors.
Introduction
Alzheimer’s disease is the most common cause of dementia among the elderly and is characterized by loss of memory and other cognitive functions. The major pathological hallmarks include extensive synaptic and neuronal loss, astrogliosis, and accumulation of proteinaceous deposits. The AD brain is characterized by the presence of β-amyloid plaques and neurofibrillary tangles (NFT), which are the hallmark pathological features (for a review see Blennow et al., 2006). β-amyloid plaques are extracellular deposits of which the major component is the β-amyloid protein (Aβ), a small polypeptide generated by processing of a much larger transmembrane β-amyloid precursor protein (APP; Masters et al., 1985; Kang et al., 1987) through the successive action of proteolytic enzymes known as secretases (for a recent review see Zhang et al., 2011). The intracellular NFT are composed of paired helical filaments of the microtubule-associated protein tau, which is abnormally hyperphosphorylated (P-tau; Grundke-Iqbal et al., 1986). Today, the focus on research has moved away from the proteinaceous deposits toward studies on the role of the triggering effectors, soluble oligomeric Aβ, and P-tau. Accordingly, much research is devoted to understanding how Aβ and P-tau lead to the toxic events associated with AD, how they cause changes in the expression of other key brain proteins and ultimately how they cause neurodegeneration. However, it is also crucial to decipher how both Aβ and P-tau interact in order to reach a better understanding of the mechanism of neurotoxicity and to achieve an effective therapy.
As extensively reviewed in this special issue, acetylcholinesterase (AChE) is a key enzyme in the cholinergic nervous system. During the progression of AD, many different types of neurons deteriorate, although there is a profound loss of forebrain cholinergic neurons, which is accompanied by a progressive decline in acetylcholine (Davies and Maloney, 1976; Perry et al., 1977). Both the acetylcholine-synthesizing enzyme choline acetyltransferase (ChAT), as well in the acetylcholine-hydrolyzing enzyme, AChE are affected. Therapies designed to reverse the cholinergic deficit are in large measure based on the importance of cholinergic function in cognition. In spite the overall decrease in the activity of AChE in the AD brain, current AD therapy is mostly based on inhibitors of AChE (AChE-I), which enhance cholinergic transmission, but which have modest and transient therapeutic effects (Giacobini, 2002; Kaduszkiewicz et al., 2005). As a consequence of its role as a target for AD therapy, AChE is one of the most studied proteins in the Alzheimer’s field, with about 1500 manuscripts indexed into the PubMed; the vast majority of reports in the field relate with treatment strategies associated with the use of AChE-I.
It has been well known for almost 50 years that the distribution of AChE molecular forms is particularly affected in the AD brain, but the physiopathological significance and subsequent implications of these intriguing changes in AChE species remain unknown. An increase in AChE levels around amyloid plaques and NFT is a common feature of AD neuropathology, and although the significance of this increase remains to be determined. In another way, up-regulation of AChE activity following long-term AChE-I therapy has been reported in a number of studies during the last decade. All these abnormalities in AChE expression patterns, as well AChE up-regulation in reaction to chronic inhibition, may are related with the limited efficiency and persistence of AChE-I. In summary, after decades of study and hundreds of reports, AChE remains of considerable interest into the AD field. The description of changes in AChE levels and forms in the AD brain has merit extensive revision (see for example Younkin et al., 1986; Mesulam and Geula, 1990; Massoulié et al., 1993; Layer, 1995; Small et al., 1996; Kása et al., 1997; Grisaru et al., 1999; Talesa, 2001; Rees and Brimijoin, 2003; Ballard et al., 2005; Silman and Sussman, 2005; Greenfield et al., 2008; Inestrosa et al., 2008; and many others). The purpose of this article is to review changes in AChE expression in the AD brain, but with a particular emphasis on the role of these changes in the pathophysiology of AD. In addition, we summarize our recent findings about the cross-talk between AChE and Aβ, and also between AChE and P-tau. The possibility that Aβ and P-tau interact through AChE is considered.
Altered AChE Molecular Form Pattern in AD
Acetylcholinesterase can exist in several different molecular forms, which have specific patterns of expression in different cell types (for a review see Massoulié, 2002). Moreover, the specific subcellular distribution of each species of AChE probably reflects different physiological functions for each form. Indeed, a large number of studies suggest that AChE could have novel functions unrelated to cholinergic neurotransmission (for review see Massoulié et al., 1993; Layer, 1995; Small et al., 1996; Soreq and Seidman, 2001). In this regard, it is important to note that AChE is present in both cholinergic and non-cholinergic brain areas, where the functional significance of non-cholinergic AChE remains unknown. We particularly refer to the work of Mesulam (2004) for a detailed view of the distribution of AChE in the non-pathological and AD brain. Even in cholinergic areas, it has been suggested that the reduction of AChE activity in the AD brain is not due to cholinergic depletion alone, as the density of AChE-rich (cholinergic) fibers decreased in cortical areas of the AD patients but was not correlated with the number of AChE-rich neurons (Heckers et al., 1992). Therefore, it is important to note that an alteration in AChE levels may not reflect a change in cholinergic neurotransmission.
Not all molecular forms of AChE are equally affected in the AD brain. Studies using sucrose gradient centrifugation have revealed two major forms of AChE in the mammal brain, tetrameric and monomeric species (Figure 1; reviewed in Massoulié et al., 1993). The major forms in the non-AD adult brain are tetramers (G4) that are anchored in the cell membrane of neurons. These tetramers probably constitute the true cholinergic species. Other minor species are monomers (G1) and dimers (G2) that cannot be completely separated from each other by sucrose gradient centrifugation. Regional variations in the AChE molecular form ratio G4/G1 usually been studied in relation to neurochemical and neuroanatomical, particularly cholinergic, features of the brain (Atack et al., 1986). However in the AD brain, there is a selective loss in the G4 form, while the lighter species are preserved (Atack et al., 1983; Fishman et al., 1986) or even increased in severely affected cases of AD (Figure 1; see also Arendt et al., 1992; Sáez-Valero et al., 1999). Similarly, changes in AChE molecular forms in cerebrospinal fluid (CSF) reflect changes in the brain (Sáez-Valero et al., 1999, 2000a). Light AChE species, which represent the major forms in plasma, are also increased in the AD plasma (García-Ayllón et al., 2010). In agreement with human studies, AChE monomeric species are also increased in brain of the APPC100 and Tg2576 transgenic mice which overproduce human Aβ (Figure 2; see also Sberna et al., 1998; Fodero et al., 2002; Silveyra et al., 2011a) and in rats given intracerebral Aβ (Sáez-Valero et al., 2002). Different reports have corroborated the possibility that Aβ might influence AChE (Sberna et al., 1997; Hu et al., 2003; Melo et al., 2003). So far, the significance of this particular increase in monomeric AChE around plaques and in Aβ models is unclear.
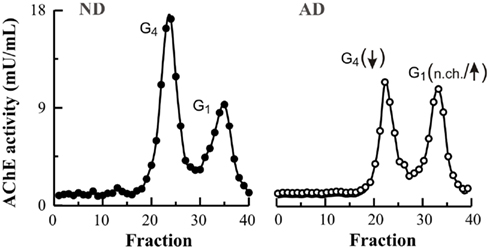
Figure 1. Tetrameric and monomeric AChE molecular forms are differentially affected in the Alzheimer’s brain. Human frontal cortex from non-demented individuals (ND; closed circles) and Alzheimer’s patients (AD; open circles) were extracted in a Tris–saline buffer containing proteinases inhibitors and 1% (w/v) Triton X-100. Molecular weight forms of AChE were analyzed by ultracentrifugation at 150,000 × g in a continuous sucrose gradient (5–20% w/v), containing 0.5% (w/v) Triton X-100, for 18 h at 4°C as previously described (Sáez-Valero et al., 1999). Tetramers (G4) and monomers (G1) AChE forms were identified by their coefficient of sedimentation. The proportion of G4 forms in AD is particularly depleted whereas the minor G1 species are mostly preserved (no change; n.ch.) or even slightly increased.
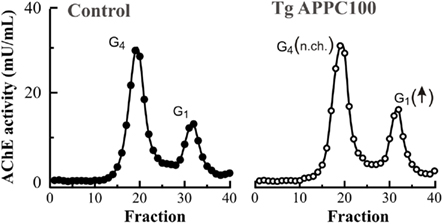
Figure 2. Monomeric AChE molecular forms are increased in the brain of a transgenic mouse model of Aβ over-expression. AChE is expressed as several different species with various molecular weights which can be identified by sucrose density gradient centrifugation (see Figure 1). Cerebral cortices from an APPC100 transgenic mice which express human Aβ (Tg APPC100; open circles) display higher levels of monomeric G1 AChE forms compared to control samples of similar age (closed circles; 3 months old), while levels of tetrameric G4 AChE species were not different (no change; n.ch.). See also Sberna et al. (1998).
Significance of Increased Monomeric AChE in AD
Light forms of AChE in the brain have been generally considered as biosynthetic precursors of the G4 forms due to the fact that oligomeric forms of AChE are assembled from monomeric precursors (Brockman et al., 1986) and that once assembled these forms do not interconvert (Rotundo, 1988). Although it is also possible that a pool of monomers represents a separate pool with a different physiological role than that of the major cholinergic G4 form (Small et al., 1996; Grisaru et al., 1999). Because G1 species are precursors of higher molecular weight species, and as a consequence G4 is always present in association with the G1 form, it is difficult to identify a specific functional pool of G1 species which is distinct from the precursor pool. Interestingly, in this context, the prevalence of lighter AChE forms in AD brain resembles an embryonic pattern of expression (Arendt et al., 1992). In the human embryonic brain, the major form of AChE is a monomeric species (Muller et al., 1985). Indeed, the expression pattern of AChE forms within the embryonic brain depends on the development stage (Zakut et al., 1985; Perry et al., 1986). The G4 species increase during human brain maturation and become the most abundant before 11 weeks of gestation (Muller et al., 1985). In rodents, the situation is similar with light AChE being the prevalent embryonic forms (Rieger and Vigny, 1976; Sung and Ruff, 1983). However, there is a shift in the molecular forms that occurs post-natally (Muller et al., 1985). The G1 AChE form is the more abundant during all gestational periods (Figure 3). Interestingly, the distribution in molecular form of the structurally related enzyme butyrylcholinesterase (BuChE) does not change significantly either during fetal development or in AD (Atack et al., 1987). The physiological significance of the early and sustained expression of embryonic G1 AChE, where a role in neurotransmission is not clear, is unknown. However, it has been suggested that AChE has roles in development, such as neuronal differentiation, regulation of cell growth, or cell adhesion. These novel functions may depend on protein–protein interactions rather than the enzyme’s catalytic activity (Brimijoin and Koenigsberger, 1999; Paraoanu et al., 2006). On this basis, it has been suggested that the AChE in AD may be similar to embryonic AChE, and that it may reflect the activation of a neuronal repair in the AD brain (Layer, 1995).
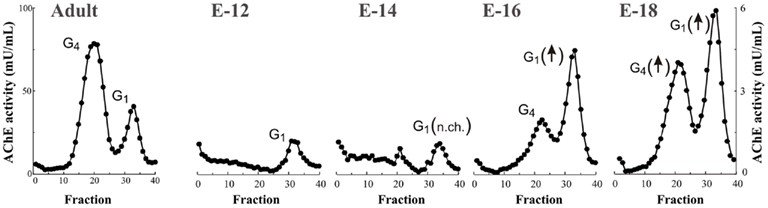
Figure 3. Monomeric AChE molecular forms are the predominant species during embryonic brain development. AChE activity was extracted from rat (Sprague-Dawley) cerebral cortex at embryonic day 12 (E14), 14 (E14), 16 (E16), and 18 (E18) and at 3 months of age (Adult), and AChE forms were separated by sucrose density gradient (see Figure 1). At early embryonic stage only light monomeric G1 AChE is present and levels are maintained (no change; n.ch.) until E15–16, an increase in G1 AChE parallels emergence of G4 AChE tetramers, the brain cholinergic species. The G4 species increase in activity during brain maturation becoming the major molecular forms during post-natal periods (N.B. difference of scales between embryonic and adult stage).
Therefore, we can speculate that G1 AChE has a non-cholinergic role during brain development, and that this role may be unrelated to the enzyme’s catalytic properties. In this context, it is assumed that all AChE forms possessed similar catalytic properties, which is probably true when oligomeric and monomeric precursors are compared. Nevertheless, it has been demonstrated subtle differences in sensitivity to inhibitors and in kinetic properties exist between tetrameric and monomeric AChE species (Ogane et al., 1992; Rakonczay, 2003). More interestingly, embryonic G4 AChE shares similar biochemical and kinetic properties with the adult enzyme, but embryonic G1 AChE differs in its kinetic properties and in its affinity for several AChE-I from the adult G1 form (Moreno et al., 1998). In this context, AChE activity present in the AD brain associated with plaques and NFT displays particular enzymatic properties and sensitivity to inhibitors (Geula and Mesulam, 1989; Wright et al., 1993).
Therefore, if we accept the possibility that embryonic AChE may possess a function independent of its catalytic capacities, the large pool of catalytically inactive AChE protein should be considered. The basis for the presence of an embryonic AChE species different from that of the adult, is unknown, but several questions arises regarding the use of AChE-I in AD therapy.
The existence of an unexpectedly large pool of inactive AChE has been demonstrated in brain (Chatel et al., 1993) and other tissues (Stieger et al., 1987; Rotundo, 1988; García-Ayllón et al., 2006), and suggested in CSF (García-Ayllón et al., 2007). This inactive pool may have non-classical functions, as it has been demonstrated that transgenic over-expression of enzymatically inactive AChE can influence neurodevelopment (Dori et al., 2005). The inactive AChE fraction is proportionally more abundant in embryonic than in adult tissues (Massoulié et al., 1993). Because routinely AChE levels are estimated enzymatically using various modifications of the Ellman et al. (1961) method, and are not normally estimated immunochemically, little information is available on this inactive pool of AChE in pathological and non-pathological conditions. A significant decrease of both AChE activity and immunoreactivity has been observed using enzyme-linked immunosorbent assay of AD brain (Hammond and Brimijoin, 1988). However, after decades of studying AChE in the AD brain further research is still necessary in order to determine the AChE protein content and its relationship with altered AChE activity levels. Whether non-catalytic AChE in brain has physiological significance, and how it is affected during pathology and treatment, are issues that warrant further study.
This complex scenario of multiple molecular forms is brought about, at least in part, by the existence of alternative splicing of the single ACHE gene, generating different AChE variants, with potential different gene regulation (Grisaru et al., 1999; see also Figure 4). Alternative 3′ RNA splicing generates different polypeptide encoding transcripts called “tailed” or T, “hydrophobic” or H, and “readthrough” or R-transcripts (Massoulié et al., 1993; Taylor and Radic, 1994; Grisaru et al., 1999), with the same catalytic domain and distinct C-terminal peptides that determine the ability of the molecule to form oligomers. In the mammalian brain, the T-transcript is the major form and encodes subunits which produce monomers, dimers, and tetrameric forms; whereas the R-transcript, which is normally present at low levels (Kaufer et al., 1998; Perrier et al., 2005), encodes a soluble monomeric form (Sternfeld et al., 2000; Figure 4). In addition to the 3′ alternatively spliced species of AChE, the 5′ end is also subject to intricate regulation (Meshorer et al., 2004) generating AChE variants that have extended N-termini; thus, within the brain N-AChE-T and N-AChE-R variants may occur in parallel with AChE-T and AChE-R. The AChE-T and AChE-R monomers or their N-extended variants cannot be distinguished by molecular weight. Whether the increase in monomeric AChE in the AD brain is related to increases in the T or R variants is still an open question. Anyhow, in this context has been suggested that the AChE-R can compete with the main brain AChE-T protein and suppress the formation of insoluble Aβ oligomers (Berson et al., 2008; see below). An increased N-AChE-T expression has been also postulated in the AD brain associated with disease progression, including apoptotic cell death (Toiber et al., 2008). In summary, Soreq and Seidman (2001) have presented evidence that N- and C-terminally modified AChE variants, all of which have similar enzymatic activities, can display distinct and in certain cases inverse functions (reviewed in Greenberg et al., 2010). The use of AChE-I which does not distinguish between AChE variants should interfere in all processes indiscriminately.
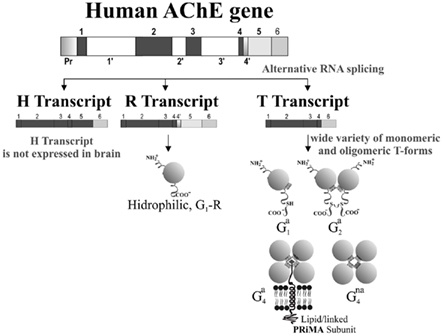
Figure 4. Diagram of the human AChE gene structure, its alternative splicing and generation of variants and molecular forms of AChE in brain. Gene structure showing the splicing pattern of the six major exons. The catalytic core of human AChE is encoded by three exons and additional exons encode the variant-specific carboxy-terminal sequences. In the normal human brain, much more AChE-T than AChE-R mRNA is produced. However, under stress AChE-R expression is increased. To date, no expression of AChE-H has been described in the brain (see Grisaru et al., 1999 for more details). The R-transcript encodes a soluble monomeric form, while the T-transcript encodes a wide variety of monomeric (G1) and oligomeric (G2 and G4) molecular forms (Massoulié et al., 1993). AChE molecular forms can vary in their solubility characteristics (non-amphiphilic: na; amphiphilic: a). The main amphiphilic G4 form in brain is anchored to the membrane by a “proline-rich membrane anchor” PRiMA subunit (Perrier et al., 2002). Additional complexity can be generated by additional 3′ alternatively splicing which can generate AChE variants with extended N-termini (not shown in figure; see Meshorer et al., 2004).
The central question is whether the changes in the distribution of AChE molecular forms in the AD brain have any physiopathological consequences. As stated previously, gross sedimentation analysis cannot distinguish between monomeric isoforms that are synthesized to be assembled in oligomers, or arise as degradation products, and those specific monomeric species which may have specific functions. Therefore, in previous studies we have further characterized the increase in monomeric AChE associated to Alzheimer’s and to Aβ by characterizing its glycosylation pattern by lectin binding analysis, based on the assumption that different functional pools of AChE may have different glycosylation patterns. Correct glycosylation determines the adequate intracellular trafficking, folding, assembly, and final localization of glycoproteins. Thus different forms (glycosylated variants or glycoforms) of the same protein should differ in glycosylation in order to achieve a different oligomerization state, subcellular localization, protein–protein interaction affinity, or a different physiological function. Indeed, for a particular glycoprotein, the abundance of single glycoforms should closely correlate to each other and be regulated within narrow limits. By exploiting the ability of lectins to bind diverse carbohydrate moieties with high specificity (Sharon and Lis, 2004), we have demonstrated that the glycosylation of AChE is altered in the AD brain, postmortem, and lumbar CSF (Sáez-Valero et al., 1997, 1999, 2000a). Tetrameric G4 and light G1 have different glycosylation patterns (Sáez-Valero et al., 1999), thus depletion specific loss of the tetrameric form in the AD brain may be responsible, in part, for this change. Changes in AChE glycosylation were also characterized in Aβ transgenic mice models displaying increases in monomeric AChE (Sberna et al., 1998; Fodero et al., 2002). However, we further demonstrated that when the light AChE species from AD and non-demented brain are isolated, G1 species present in AD brain displayed different affinities for lectins and for conformational anti-AChE antibodies, compared with isoforms from control brains (Sáez-Valero et al., 2000b). These changes indicate that the minor subset of G1 AChE, whose contribution is increased in AD brain, correspond to isoforms (glycoforms) either not present, or poorly present in adult human brain in non-disease conditions. The physiological relevance of the increase in this minor G1 form for AD pathogenesis is unclear.
Alterations in the glycosylation state of other glycoproteins have been reported in AD tissue (Guevara et al., 1998; Fodero et al., 2001; Kanninen et al., 2004; Sihlbom et al., 2008). Pathological impairment in the broader protein glycosylation machinery could significantly compromise the processing of many glycoproteins, thereby resulting in loss of physiological function of many of these proteins. Abnormal incorporation of carbohydrate moieties in AChE subunits can compromise its functional role and/or oligomerization. In this context, altered AChE glycosylation has been also demonstrated in Creutzfeldt-Jakob disease (Silveyra et al., 2006), a neurodegenerative process where impaired glycosylation machinery is suspected (Rudd et al., 1999). Nonetheless, unlike AD, in Creutzfeldt-Jakob disease, altered AChE glycosylation is not caused by changes in the proportion of any particular molecular form (Silveyra et al., 2006). Therefore, we favor the hypothesis that in Creutzfeldt-Jakob disease change in AChE glycosylation is the consequence of perturbed glycosylation machinery, whereas the altered glycosylation pattern of light AChE species in AD reflects an imbalance of protein glycoforms resulting from changes in AChE variants related or not to differentiation state. This is an issue that requires further study.
P-Tau Increases AChE, Differences from Aβ
It has been noted that abnormal AChE expression in the AD brain occurs in association with the two hallmark features of the AD pathology, the amyloid plaques and the NFT (Mesulam and Morán, 1987; Ulrich et al., 1990). As mentioned previously, Aβ peptides influence AChE levels, thus Aβ may be responsible for increased AChE around plaques. However, the increase in AChE associated with NFT has remained largely unexplored. Recently, we showed for the first time that P-tau can trigger an increase in AChE expression (Silveyra et al., 2011a). In vivo over-expression of P-tau in transgenic mice (Tg VLW mice) expressing human tau mutations causing frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) led to an increase in the activity of the T-transcript of AChE (Silveyra et al., 2011a). The results suggest that the early increase in AChE expression that occurs around NFT may be a consequence of disturbed tau phosphorylation. In contrast to Aβ transgenic models in which only one specific molecular form of AChE increases, in the P-tau transgenic mice, all major molecular forms of AChE were increased, including the tetrameric species (Figure 5). The explanation for differences in the pattern of AChE expression between Aβ and P-tau over-expressing mice remain unclear and will require further research, as will the potential differing physiopathological consequences of increases in tetrameric or monomeric AChE. In this context, it is necessary to consider the different subcellular localization of tetramers and monomers. Light forms probably localized internally whereas G4 forms are localized extracellularly (Inestrosa et al., 1981). Therefore, the different localization of these forms determines and limits potential interactions with other protein partners. The recent finding that PRiMA (proline-rich membrane anchor) directs a restricted localization of tetrameric AChE not only to synapses but also to membrane microdomains called rafts (Xie et al., 2010), suggests that there may be different functional interactions of AChE species with particular partners.
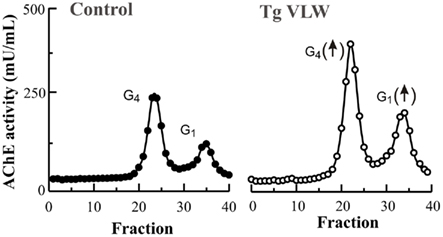
Figure 5. All molecular forms of AChE are increased in the brain of a transgenic mice model of P-tau over-expression. In a transgenic mice (Tg VLW; open circles) expressing human tau mutations causing frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), both the major cholinergic tetramers G4 AChE and light monomeric forms G1 AChE activities were higher compared to age-matched control-littermate mice (closed circles; 5 months old).
Downstream Consequences of Changes in AChE Levels in AD Brain
Notwithstanding the overall loss of total AChE activity in the AD brain, AChE is consistently increased in regions around amyloid plaques and NFT at all stages of the disease, including some of the earliest stages (Perry et al., 1980; Mesulam and Morán, 1987; Ulrich et al., 1990). Extensive studies by Inestrosa et al. (1996) suggest that AChE may directly interact with Aβ in a manner that increases deposition of Aβ to form plaques. Studies using double transgenic mice that over-express AChE and Aβ (Rees et al., 2003) support these observations and they suggest that AChE may play a role in pathogenesis of AD.
Our own recent work also indicate that AChE can modulate APP processing and Aβ production. Aβ is produced through the successive action of two proteolytic enzymes, β-secretase and Γ-secretase on APP (Zhang et al., 2011). The active proteolytic component of the Γ-secretase complex is presenilin 1 (PS1; Suh and Checler, 2002). Mutations in PS1 cause early-onset AD with an accelerated rate of Aβ deposition (St George-Hyslop, 2000), thus proteins that interact with PS1 are of major functional importance. We have previously demonstrated an interaction between AChE and PS1 by reciprocal co-immunoprecipitation (Silveyra et al., 2008). Recently, we demonstrated that AChE can influence PS1 levels by showing that AChE over-expression increases PS1 levels, while AChE knock-down with siRNA leads decrease PS1 in transfected cells (Silveyra et al., 2011b). Perhaps the most significant conclusion from our recent study is the potential participation of AChE in a degenerative cycle that enhances amyloidogenic APP processing. We can presume that several degenerative cycles, participating as interactive systems within a larger vicious cycle, accelerate the development of AD. Hence, in AD it is possible that Aβ can induce a feedback loop leading to amyloidogenic APP processing (Cribbs et al., 1995; White et al., 2003). A vicious cycle of Aβ generation potentially could involve PS1; recent evidence indicates that Aβ42 can induce an increase in PS1 levels in cultured neurons creating a toxic loop (Matrone et al., 2008). Using different experimental conditions, we have confirmed that both Aβ42 and P-tau trigger an increase in AChE, which can in turn influence PS1 and thereby modulate Aβ production (Figure 6). We have found that Aβ42-induced PS1 increase can be prevented by pre-treatment of SH-SY5Y cells with siRNA AChE (Silveyra et al., 2011b). The possibility that different effects may be obtained with different AChE species and variants (tetrameric versus monomeric molecular form, or T-variant versus R-variant or N-extended variant), requires further study. In this context, recent evidence indicates a specific role inducing GSK-3β activation and tau hyperphosphorylation for the alternative N-terminally extended T-AChE variant (Toiber et al., 2008), variant up-regulated by stressors inducing protein misfolding and calcium imbalances, both characteristic of AD.
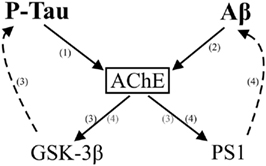
Figure 6. Schematic representation of the multiple relationships between AChE and the Alzheimer’s effectors Aβ and P-tau. AChE is proposed to interact with both Aβ and P-tau. P-tau can lead to an increase in the activity of AChE (1) (Silveyra et al., 2011a), whereas over-expression of N-terminally extended T-AChE variant activates the tau kinase GSK-3β inducing tau hyperphosphorylation (3) (Toiber et al., 2008). In addition, Aβ peptides can also increase AChE levels (2) (Sberna et al., 1997). In turn, AChE can affect APP processing and Aβ production by modulating the levels of the Γ-secretase catalytic subunit PS1 (4) (Silveyra et al., 2011b). Thus, increased AChE can affect both amyloidogenic and tau hyperphosphorylation pathways.
It appears likely, therefore, that several vicious cycles trigger by Aβ and P-tau involve the potential participation of AChE. The significance of these theoretical considerations to the clinical and neuropathological course of AD remains to be demonstrated.
Effects of AChE-I
Therapies designed to reverse the cholinergic deficit are in large measure based on the importance of cholinergic function in cognition. Indeed, AChE-Is have proven to be modestly efficacious in treating the cognitive and functional symptoms of AD. In addition disease-modifying effects of AChE inhibition has been also considered (Giacobini, 2002). However, the modest clinical efficacy of AChE-Is has not discouraged the development of new AChE-Is, and particularly the so called dual binding site AChE-Is (Muñoz-Torrero, 2008; Pepeu and Giovannini, 2009), which are both inhibitors of AChE and also of amyloid plaque formation. This latter effect is based on the fact that AChE-binding molecules may, in addition to their effect on enzyme activity, block the effect of AChE on Aβ fibrillogenesis by interaction throughout the peripheral anionic site of the enzyme (Inestrosa et al., 1996). Besides the design of new inhibitors with the capacity to block catalytic and peripheral anionic sites of AChE, there is evidence that inhibitors may also influence APP processing. AChE-Is have been shown to alter APP expression and metabolism in cellular (Lahiri et al., 1994; Pakaski et al., 2001; Peng et al., 2006) and animal models (Mori et al., 1995; Zimmermann et al., 2004; Dong et al., 2009), as well in AD-treated patients (Clarke et al., 2001; Basun et al., 2002; Zimmermann et al., 2005). The modulatory effects of AChE-I on APP metabolism have been attributed to their effect on ADAM10/α-secretase (Zimmermann et al., 2004; Peng et al., 2006), BACE1/β-secretase (Lahiri et al., 2007; Fu et al., 2008; Li et al., 2010), and on PS1/Γ-secretase (Silveyra et al., 2011b). The disparity between the effects of AChE-Is and cholinergic agonists on secretase activity would suggest that AChE is not exerting its modulatory action on PS1 via a cholinergic mechanism (Zimmermann et al., 2004; Silveyra et al., 2011b). However, effects on APP processing by cholinergic agonists have been also demonstrated (Nitsch et al., 1992; Rossner et al., 1998; Davis et al., 2010). The mechanisms by which AChE-Is influence APP processing remain unclear, but it is suggested that regulation of APP processing by AChE-Is may involve multiple mechanisms, including cholinergic and non-cholinergic actions, some independent of their anti-cholinesterase (catalytic) activities (Lahiri et al., 1997).
The therapeutic effect of current AChE-Is is both modest and transient. Current AChE-I treatment results are disappointing both because of their poor efficacy and tolerability. Interestingly, these drugs have a limited duration of cognitive benefit. The effects of these drugs on APP processing also fail to be maintained over the long-term in Alzheimer’s patients (Basun et al., 2002).
The transience of the response to AChE-I could be associated with AChE up-regulation in reaction to chronic inhibition (Chiappa et al., 1995; Kaufer et al., 1998). Indeed, increases in CSF AChE have been reported after AChE-I treatment (Davidsson et al., 2001; Darreh-Shori et al., 2006; García-Ayllón et al., 2007; Parnetti et al., 2011). The varying responses of different AChE species to AChE-I treatment, were the light AChE species seem not subject to AChE-I induced up-regulation, suggest different modes of regulation and should be also considered (García-Ayllón et al., 2007). Interestingly, we found that the effect of AChE-I treatment on PS1 levels was also not sustained, and that the lack of effect on PS1 was associated with up-regulation of AChE (Silveyra et al., 2011b). Our data suggest that sustained AChE inhibition cannot be effective when the expression of AChE is up-regulated and that this undesired effect needs to be addressed to develop more effective therapies based on AChE-I.
Summary and Conclusion
In summary, AChE species differ in their responses to disease and their interactions with β-amyloid and P-tau. The important question about the nature of the alternative functions of AChE, their association with different AChE species and variants, and their role in AD pathogenesis and therapy needs to be examined further. Recent evidence also suggests the potential participation of AChE in vicious cycles involving Aβ and P-tau. Elucidation of the mechanisms involved in these changes will be useful for understanding the physiological and pathological relevance of altered AChE expression in the AD brain and AChE-I pharmacological intervention. The chronic increases in AChE activity during AChE-I treatment may cause the therapeutic value of AChE-I to be limited and temporary and needs to be addressed in order to improve therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would especially like to thank Dr. M.-X. Silveyra for her contribution, and M.-T. García-Hedo and C. Serra-Basante for technical assistance. This work was supported by grants from Foundatión CIEN-Reina Sofía, FIS (Grant PS09/00684), and CIBERNED, ISC-III from Spain to Javier Sáez-Valero.
References
Arendt, T., Bruckner, M. K., Lange, M., and Bigl, V. (1992). Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development – a study of molecular forms. Neurochem. Int. 21, 381–396.
Atack, J. R., Perry, E. K., Bonham, J. R., Candy, J. M., and Perry, R. H. (1987). Molecular forms of butyrylcholinesterase in the human neocortex during development and degeneration of the cortical cholinergic system. J. Neurochem. 48, 1687–1692.
Atack, J. R., Perry, E. K., Bonham, J. R., Candy, J. M., and Perry, R. H. (1986). Molecular forms of acetylcholinesterase and butyrylcholinesterase in the aged human central nervous system. J. Neurochem. 47, 263–277.
Atack, J. R., Perry, E. K., Bonham, J. R., Perry, R. H., Tomlinson, B. E., Blessed, G., and Fairbairn, A. (1983). Molecular forms of acetylcholinesterase in senile dementia of Alzheimer type: selective loss of the intermediate (10S) form. Neurosci. Lett. 40, 199–204.
Ballard, C. G., Greig, N. H., Guillozet-Bongaarts, A. L., Enz, A., and Darvesh, S. (2005). Cholinesterases: roles in the brain during health and disease. Curr. Alzheimer Res. 2, 307–318.
Basun, H., Nilsberth, C., Eckman, C., Lannfelt, L., and Younkin, S. (2002). Plasma levels of Abeta42 and Abeta40 in Alzheimer patients during treatment with the acetylcholinesterase inhibitor tacrine. Dement. Geriatr. Cogn. Disord. 14, 156–160.
Berson, A., Knobloch, M., Hanan, M., Diamant, S., Sharoni, M., Schuppli, D., Geyer, B. C., Ravid, R., Mor, T. S., Nitsch, R. M., and Soreq, H. (2008). Changes in readthrough acetylcholinesterase expression modulate amyloid-beta pathology. Brain 131, 109–119.
Brimijoin, S., and Koenigsberger, C. (1999). Cholinesterases in neural development: new findings and toxicologic implications. Environ. Health Perspect. 107, 59–64.
Brockman, S. K., Usiak, M. F., and Younkin, S. G. (1986). Assembly of monomeric acetylcholinesterase into tetrameric and asymmetric forms. J. Biol. Chem. 261, 1201–1207.
Chatel, J. M., Grassi, J., Frobert, Y., Massoulié, J., and Vallette, F. M. (1993). Existence of an inactive pool of acetylcholinesterase in chicken brain. Proc. Natl. Acad. Sci. U.S.A. 90, 2476–2480.
Chiappa, S., Padilla, S., Koenigsberger, C., Moser, V., and Brimijoin, S. (1995). Slow accumulation of acetylcholinesterase in rat brain during enzyme inhibition by repeated dosing with chlorpyrifos. Biochem. Pharmacol. 49, 955–963.
Clarke, N. A., Soininen, H., Gustafson, L., Minthon, L., Alhainen, K., and Francis, P. T. (2001). Tacrine may alter APP-like protein levels in the lumbar CSF of Alzheimer patients. Int. J. Geriatr. Psychiatry 16, 1104–1106.
Cribbs, D. H., Davis-Salinas, J., Cotman, C. W., and Van Nostrand, W. E. (1995). Aß induces increased expression and processing of amyloid precursor protein in cortical neurons. Alzheimer Res. 1, 197–200.
Darreh-Shori, T., Meurling, L., Pettersson, T., Hugosson, K., Hellström-Lindahl, E., Andreasen, N., Minthon, L., and Nordberg, A. (2006). Changes in the activity and protein levels of CSF acetylcholinesterases in relation to cognitive function of patients with mild Alzheimer’s disease following chronic donepezil treatment. J. Neural Transm. 113, 1791–1801.
Davidsson, P., Blennow, K., Andreasen, N., Eriksson, B., Minthon, L., and Hesse, C. (2001). Differential increase in cerebrospinal fluid-acetylcholinesterase after treatment with acetylcholinesterase inhibitors in patients with Alzheimer’s disease. Neurosci. Lett. 300, 157–160.
Davies, P., and Maloney, A. J. F. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2, 1403.
Davis, A. A., Fritz, J. J., Wess, J., Lah, J. J., and Levey, A. I. (2010). Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J. Neurosci. 30, 4190–4196.
Dong, H., Yuede, C. M., Coughlan, C. A., Murphy, K. M., and Csernansky, J. G. (2009). Effects of donepezil on amyloid-beta and synapse density in the Tg2576 mouse model of Alzheimer’s disease. Brain Res. 1303, 169–178.
Dori, A., Cohen, J., Silverman, W. F., Pollack, Y., and Soreq, H. (2005). Functional manipulations of acetylcholinesterase splice variants highlight alternative splicing contributions to murine neocortical development. Cereb. Cortex 15, 419–430.
Ellman, G. L., Courtney, K. D., Andres, V. Jr., and Feather-Stone, R. M. (1961). A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95.
Fishman, E. B., Siek, G. C., MacCallum, R. D., Bird, E. D., Volicer, L., and Marquis, J. K. (1986). Distribution of the molecular forms of acetylcholinesterase in human brain, alterations in dementia of the Alzheimer type. Ann. Neurol. 19, 246–252.
Fodero, L. R., Sáez-Valero, J., Barquero, M. S., Marcos, A., McLean, C. A., and Small, D. H. (2001). Wheat germ agglutinin-binding glycoproteins are decreased in Alzheimer’s disease cerebrospinal fluid. J. Neurochem. 79, 1022–1006.
Fodero, L. R., Sáez-Valero, J., McLean, C. A., Martins, R. N., Beyreuther, K., Masters, C. L., Robertson, T. A., and Small, D. H. (2002). Altered glycosylation of acetylcholinesterase in APP (SW) Tg2576 transgenic mice occurs prior to amyloid plaque deposition. J. Neurochem. 81, 441–448.
Fu, H., Li, W., Luo, J., Lee, N. T., Li, M., Tsim, K. W., Pang, Y., Youdim, M. B., and Han, Y. (2008). Promising anti-Alzheimer’s dimer bis(7)-tacrine reduces beta-amyloid generation by directly inhibiting BACE-1 activity. Biochem. Biophys. Res. Commun. 366, 631–636.
García-Ayllón, M. S., Riba-Llena, I., Serra-Basante, C., Alom, J., Boopathy, R., and Sáez-Valero, J. (2010). Altered levels of acetylcholinesterase in Alzheimer plasma. PLoS ONE 5, e8701.
García-Ayllón, M. S., Silveyra, M. X., Andreasen, N., Brimijoin, S., Blennow, K., and Sáez-Valero, J. (2007). Cerebrospinal fluid acetylcholinesterase changes after treatment with donepezil in patients with Alzheimer’s disease. J. Neurochem. 101, 1701–1711.
García-Ayllón, M. S., Silveyra, M. X., Candela, A., Compañ, A., Clària, J., Jover, R., Pérez-Mateo, M., Felipo, V., Martínez, S., Galcerán, J., and Sáez-Valero, J. (2006). Changes in liver and plasma acetylcholinesterase in rats with cirrhosis induced by bile duct ligation. Hepatology 43, 444–453.
Geula, C., and Mesulam, M.-M. (1989). Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res. 498, 185–189.
Giacobini, E. (2002). Long-term stabilizing effect of cholinesterase inhibitors in the therapy of Alzheimer’ disease. J. Neural Transm. Suppl. 62, 181–187.
Greenberg, D. S., Toiber, D., Berson, A., and Soreq, H. (2010). Acetylcholinesterase variants in Alzheimer’s disease: from neuroprotection to programmed cell death. Neurodegener. Dis. 7, 60–63.
Greenfield, S. A., Zimmermann, M., and Bond, C. E. (2008). Non-hydrolytic functions of acetylcholinesterase. The significance of C-terminal peptides. FEBS J. 275, 604–611.
Grisaru, D., Sternfeld, M., Eldor, A., Glick, D., and Soreq, H. (1999). Structural roles of acetylcholinesterase variants in biology and pathology. Eur. J. Biochem. 264, 672–686.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4907–4913.
Guevara, J., Espinosa, B., Zenteno, E., Vázguez, L., Luna, J., Perry, G., and Mena, R. (1998). Altered glycosylation pattern of proteins in Alzheimer disease. J. Neuropathol. Exp. Neurol. 57, 905–914.
Hammond, P., and Brimijoin, S. (1988). Acetylcholinesterase in Huntington’s and Alzheimer’s diseases: simultaneous enzyme assay and immunoassay of multiple brain regions. J. Neurochem. 50, 1111–1116.
Heckers, S., Geula, C., and Mesulam, M. M. (1992). Acetylcholinesterase-rich pyramidal neurons in Alzheimer’s disease. Neurobiol. Aging 13, 455–460.
Hu, W., Gray, N. W., and Brimijoin, S. (2003). Amyloid-beta increases acetylcholinesterase expression in neuroblastoma cells by reducing enzyme degradation. J. Neurochem. 86, 470–478.
Inestrosa, N. C., Alvarez, A., Perez, C. A., Moreno, R. D., Vicente, M., Linker, C., Casanueva, O. I., Soto, C., and Garrido, J. (1996). Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron 16, 881–891.
Inestrosa, N. C., Dinamarca, M. C., and Alvarez, A. (2008). Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 275, 625–632.
Inestrosa, N. C., Reiness, C. G., Reichardt, L. F., and Hall, Z. W. (1981). Cellular localization of the molecular forms of acetylcholinesterase in rat pheochromocytoma PC12 cells treated with nerve growth factor. J. Neurosci. 1, 1260–1267.
Kaduszkiewicz, H., Zimmermann, T., Beck-Bornholdt, H. P., and van den Bussche, H. (2005). Cholinesterase inhibitors for patients with Alzheimer’s disease: systematic review of randomised clinical trials. BMJ 331, 321–327.
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., Multhaup, G., Beyreuther, K., and Müller-Hill, B. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 19–25; 325, 733.
Kanninen, K., Goldsteins, G., Auriola, S., Alafuzoff, I., and Koistinaho, J. (2004). Glycosylation changes in Alzheimer’s disease as revealed by a proteomic approach. Neurosci. Lett. 367, 235–240.
Kása, P., Rakonczay, Z., and Gulya, K. (1997). The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 52, 511–535.
Kaufer, D., Friedman, A., Seidman, S., and Soreq, H. (1998). Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 393, 373–377.
Lahiri, D. K., Chen, D., Maloney, B., Holloway, H. W., Yu, Q. S., Utsuki, T., Giordano, T., Sambamurti, K., and Greig, N. H. (2007). The experimental Alzheimer’s disease drug posiphen [(+)-phenserine] lowers amyloid-beta peptide levels in cell culture and mice. J. Pharmacol. Exp. Ther. 320, 386–396.
Lahiri, D. K., Farlow, M. R., Nurnberger, J. I. Jr., and Greig, N. H. (1997). Effects of cholinesterase inhibitors on the secretion of beta-amyloid precursor protein in cell cultures. Ann. N. Y. Acad. Sci. 826, 416–421.
Lahiri, D. K., Lewis, S., and Farlow, M. R. (1994). Tacrine alters the secretion of the beta-amyloid precursor protein in cell lines. J. Neurosci. Res. 37, 777–787.
Layer, P. G. (1995). Nonclassical roles of cholinesterases in the embryonic brain and possible links to Alzheimer disease. Alzheimer Dis. Assoc. Disord. 9, 29–36.
Li, Q., Wu, D., Zhang, L., and Zhang, Y. (2010). Effects of galantamine on ß-amyloid release and beta-site cleaving enzyme 1 expression in differentiated human neuroblastoma SH-SY5Y cells. Exp. Gerontol. 45, 842–847.
Massoulié, J. (2002). The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals 11, 130–143.
Massoulié, J., Pezzementi, L., Bon, S., Krejci, E., and Vallette, F.-M. (1993). Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 41, 31–91.
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer’s disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249.
Matrone, C., Ciotti, M. T., Mercanti, D., Marolda, R., and Calissano, P. (2008). NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 13139–11344.
Melo, J. B., Agostinho, P., and Oliveira, C. R. (2003). Involvement of oxidative stress in the enhancement of acetylcholinesterase activity induced by amyloid beta-peptide. Neurosci. Res. 45, 117–127.
Meshorer, E., Toiber, D., Zurel, D., Sahly, I., Dori, A., Cagnano, E., Schreiber, L., Grisaru, D., Tronche, F., and Soreq, H. (2004). Combinatorial complexity of 5′ alternative acetylcholinesterase transcripts and protein products. J. Biol. Chem. 279, 29740–29751.
Mesulam, M. M. (2004). The cholinergic innervation of the human cerebral cortex. Prog. Brain Res. 145, 67–78.
Mesulam, M. M., and Geula, C. (1990). Shifting patterns of cortical cholinesterases in Alzheimer’s disease: implications for treatment, diagnosis, and pathogenesis. Adv. Neurol. 51, 235–240.
Mesulam, M. M., and Morán, M. A. (1987). Cholinesterases within neurofibrillary tangles related to age and Alzheimer’s disease. Ann. Neurol. 22, 223–228.
Moreno, R. D., Campos, F. O., Dajas, F., and Inestrosa, N. C. (1998). Developmental regulation of mouse brain monomeric acetylcholinesterase. Int. J. Dev. Neurosci. 16, 123–134.
Mori, F., Lai, C. C., Fusi, F., and Giacobini, E. (1995). Cholinesterase inhibitors increase secretion of APPs in rat brain cortex. Neuroreport 6, 633–636.
Muller, F., Dumez, Y., and Massoulié, J. (1985). Molecular forms and solubility of acetylcholinesterase during the embryonic development of rat and human brain. Brain Res. 331, 295–302.
Muñoz-Torrero, D. (2008). Acetylcholinesterase inhibitors as disease-modifying therapies for Alzheimer’s disease. Curr. Med. Chem. 15, 2433–2455.
Nitsch, R. M., Slack, B. E., Wurtman, R. J., and Growdon, J. H. (1992). Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258, 304–307.
Ogane, N., Giacobini, E., and Messamore, E. (1992). Preferential inhibition of acetylcholinesterase molecular forms in rat brain. Neurochem. Res. 17, 489–495.
Pakaski, M., Papp, H., Rakonczay, Z., Fakla, I., and Kasa, P. (2001). Effects of acetylcholinesterase inhibitors on the metabolism of amyloid precursor protein in vitro. Neurobiology (Bp.) 9, 55–57.
Paraoanu, L. E., Steinert, G., Klaczinski, J., Becker-Röck, M., Bytyqi, A., and Layer, P. G. (2006). On functions of cholinesterases during embryonic development. J. Mol. Neurosci. 30, 201–204.
Parnetti, L., Chiasserini, D., Andreasson, U., Ohlson, M., Hüls, C., Zetterberg, H., Minthon, L., Wallin, A. K., Andreasen, N., Talesa, V. N., and Blennow, K. (2011). Changes in CSF acetyl- and butyrylcholinesterase activity after long-term treatment with AChE inhibitors in Alzheimer’s disease. Acta Neurol. Scand. 124, 122–129.
Peng, Y., Jiang, L., Lee, D. Y., Schachter, S. C., Ma, Z., and Lemere, C. A. (2006). Effects of huperzine a on amyloid precursor protein processing and beta-amyloid generation in human embryonic kidney 293 APP Swedish mutant cells. J. Neurosci. Res. 84, 903–911.
Pepeu, G., and Giovannini, M. G. (2009). Cholinesterase inhibitors and beyond. Curr. Alzheimer Res. 6, 86–96.
Perrier, A. L., Massoulié, J., and Krejci, E. (2002). PRiMA: the membrane anchor of acetylcholinesterase in the brain. Neuron 33, 275–285.
Perrier, N. A., Salani, M., Falasca, C., Bom, S., Augusti-Tocco, G., and Massoulié, J. (2005). The read through variant of acetylcholinesterase remains very minor after heat shock, organophosphate inhibition and stress, in cell culture and in vivo. J. Neurochem. 94, 629–638.
Perry, R. H., Blessed, G., Perry, E. K., and Tomlinson, B. E. (1980). Histochemical observations on cholinesterase activities in the brains of elderly normal and demented (Alzheimer-type) patients. Age Ageing 9, 9–16.
Perry, E. K., Perry, R. H., Blessed, G., and Tomlinson, B. E. (1977). Necropsy evidence of central cholinergic deficits in senile dementia. Lancet 1, 189.
Perry, E. K., Smith, C. J., Atack, J. R., Candy, J. M., Johnson, M., and Perry, R. H. (1986). Neocortical cholinergic enzyme and receptor activities in the human fetal brain. J. Neurochem. 47, 1262–1269.
Rakonczay, Z. (2003). Potencies and selectivities of inhibitors of acetylcholinesterase and its molecular forms in normal and Alzheimer’s disease brain. Acta Biol. Hung. 54, 183–189.
Rees, T., Hammond, P. I., Soreq, H., Younkin, S., and Brimijoin, S. (2003). Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiol. Aging 24, 777–787.
Rees, T. M., and Brimijoin, S. (2003). The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. Drugs Today (Barc.) 39, 75–83.
Rieger, F., and Vigny, M. (1976). Solubilization and physicochemical characterization of rat brain acetylcholinesterase: development and maturation of its molecular forms. J. Neurochem. 27, 121–129.
Rossner, S., Ueberham, U., Schliebs, R., Perez-Polo, J. R., and Bigl, V. (1998). The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog. Neurobiol. 56, 541–569.
Rotundo, R. L. (1988). Biogenesis of acetylcholinesterase molecular forms in muscle. Evidence for a rapidly turning over, catalytically inactive precursor pool. J. Biol. Chem. 263, 19398–19406.
Rudd, P. M., Endo, T., Colominas, C., Groth, D., Wheeler, S. F., Harvey, D. J., Wormald, M. R., Serban, H., Prusiner, S. B., Kobata, A., and Dwek, R. A. (1999). Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl. Acad. Sci. U.S.A. 96, 13044–13049.
Sáez-Valero, J., Barquero, M. S., Marcos, A., McLean, C. A., and Small, D. H. (2000a). Altered glycosylation of acetylcholinesterase in lumbar cerebrospinal fluid of patients with Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatr. 69, 664–667.
Sáez-Valero, J., Mok, S. S., and Small, D. H. (2000b). An unusually glycosylated form of acetylcholinesterase is a CSF biomarker for Alzheimer’s disease. Acta Neurol. Scand. Suppl. 176, 49–52.
Sáez-Valero, J., de Ceballos, M. L., Small, D. H., and de Felipe, C. (2002). Changes in molecular isoform distribution of acetylcholinesterase in rat cortex and cerebrospinal fluid after intracerebroventricular administration of amyloid beta-peptide. Neurosci. Lett. 325, 199–202.
Sáez-Valero, J., Sberna, G., McLean, C. A., Masters, C. L., and Small, D. H. (1997). Glycosylation of acetylcholinesterase as diagnostic marker for Alzheimer’s disease. Lancet 350, 929.
Sáez-Valero, J., Sberna, G., McLean, C. A., and Small, D. H. (1999). Molecular isoform distribution and glycosylation of acetylcholinesterase are altered in brain and cerebrospinal fluid of patients with Alzheimer’s disease. J. Neurochem. 72, 1600–1608.
Sberna, G., Sáez-Valero, J., Beyreuther, K., Masters, C. L., and Small, D. H. (1997). The amyloid β-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J. Neurochem. 69, 1177–1184.
Sberna, G., Sáez-Valero, J., Li, Q. X., Czech, C., Beyreuther, K., Masters, C. L., McLean, C. A., and Small, D. H. (1998). Acetylcholinesterase is increased in the brains of transgenic mice expressing the C-terminal fragment (CT100) of the beta-amyloid protein precursor of Alzheimer’s disease. J. Neurochem. 71, 723–731.
Sharon, N., and Lis, H. (2004). History of lectins: from hemagglutinins to biological recognition molecules. Glycobiology 14, 53R–62R.
Sihlbom, C., Davidsson, P., Sjögren, M., Wahlund, L. O., and Nilsson, C. L. (2008). Structural and quantitative comparison of cerebrospinal fluid glycoproteins in Alzheimer’s disease patients and healthy individuals. Neurochem. Res. 33, 1332–1340.
Silman, I., and Sussman, J. L. (2005). Acetylcholinesterase: “classical” and “non-classical” functions and pharmacology. Curr. Opin. Pharmacol. 5, 293–302.
Silveyra, M. X., Cuadrado-Corrales, N., Marcos, A., Barquero, M. S., Rábano, A., Calero, M., and Sáez-Valero, J. (2006). Altered glycosylation of acetylcholinesterase in Creutzfeldt-Jakob disease. J. Neurochem. 96, 97–104.
Silveyra, M. X., Evin, G., Montenegro, M. F., Vidal, C. J., Martínez, S., Culvenor, J. G., and Sáez-Valero, J. (2008). Presenilin 1 interacts with acetylcholinesterase and alters its enzymatic activity and glycosylation. Mol. Cell Biol. 28, 2908–2919.
Silveyra, M. X., García-Ayllón, M. S., de Barreda, E. G., Small, D. H., Martínez, S., Avila, J., and Sáez-Valero, J. (2011a). Altered expression of brain acetylcholinesterase in FTDP-17 human tau transgenic mice. Neurobiol. Aging. [Epub ahead of print]
Silveyra, M. X., García-Ayllón, M. S., Serra-Basante, C., Mazzoni, V., García-Gutierrez, M. S., Manzanares, J., Culvenor, J. G., and Sáez-Valero, J. (2011b). Changes in acetylcholinesterase expression are associated with altered presenilin-1 levels. Neurobiol. Aging. [Epub ahead of print].
Small, D. H., Michaelson, S., and Sberna, G. (1996). Non-classical actions of cholinesterases: role in cellular differentiation, tumorigenesis and Alzheimer’s disease. Neurochem. Int. 28, 453–483.
Soreq, H., and Seidman, S. (2001). Acetylcholinesterase – new roles for an old actor. Nat. Rev. Neurosci. 2, 294–302.
St George-Hyslop, P. H. (2000). Molecular genetics of Alzheimer’s disease. Biol. Psychiatry 47, 183–199.
Sternfeld, M., Shoham, S., Klein, O., Flores-Flores, C., Evron, T., Idelson, G. H., Kitsberg, D., Patrick, J. W., and Soreq, H. (2000). Excess “read-through” acetylcholinesterase attenuates but the “synaptic” variant intensifies neurodeterioration correlates. Proc. Natl. Acad. Sci. U.S.A. 97, 8647–8652.
Stieger, S., Brodbeck, U., and Witzemann, V. (1987). Inactive monomeric acetylcholinesterase in the low-salt-soluble extract of the electric organ from Torpedo marmorata. J. Neurochem. 49, 460–467.
Suh, Y. H., and Checler, F. (2002). Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol Rev. 54, 469–525.
Sung, S. C., and Ruff, B. A. (1983). Molecular forms of sucrose extractable and particulate acetylcholinesterase in the developing and adult rat brain. Neurochem. Res. 8, 303–311.
Talesa, V. N. (2001). Acetylcholinesterase in Alzheimer’s disease. Mech. Ageing Dev. 122, 1961–1969.
Taylor, P., and Radic, Z. (1994). The cholinesterases: from genes to proteins. Annu. Rev. Pharmacol. Toxicol. 34, 281–320.
Toiber, D., Berson, A., Greenberg, D., Melamed-Book, N., Diamant, S., and Soreq, H. (2008). N-acetylcholinesterase-induced apoptosis in Alzheimer’s disease. PLoS ONE 3, e3108.
Ulrich, J., Meier-Ruge, W., Probst, A., Meier, E., and Ipsen, S. (1990). Senile plaques: staining for acetylcholinesterase and A4 protein: a comparative study in the hippocampus and entorhinal cortex. Acta Neuropathol. 80, 624–628.
White, A. R., Maher, F., Brazier, M. W., Jobling, M. F., Thyer, J., Stewart, L. R., Thompson, A., Gibson, R., Masters, C. L., Multhaup, G., Beyreuther, K., Barrow, C. J., Collins, S. J., and Cappai, R. (2003). Diverse fibrillar peptides directly bind the Alzheimer’s amyloid precursor protein and amyloid precursor-like protein 2 resulting in cellular accumulation. Brain Res. 966, 231–244.
Wright, C. I., Geula, C., and Mesulam, M.-M. (1993). Neuroglial cholinesterases in the normal brain and in Alzheimer’s disease, relationship to plaques, tangles and patterns of selective vulnerability. Ann. Neurol. 34, 373–384.
Xie, H. Q., Liang, D., Leung, K. W., Chen, V. P., Zhu, K. Y., Chan, W. K., Choi, R. C., Massoulié, J., and Tsim, K. W. (2010). Targeting acetylcholinesterase to membrane rafts: a function mediated by the proline-rich membrane anchor (PRiMA) in neurons. J. Biol. Chem. 285, 11537–11546.
Younkin, S. G., Goodridge, B., Katz, J., Lockett, G., Nafziger, D., Usiak, M. F., and Younkin, L. H. (1986). Molecular forms of acetylcholinesterases in Alzheimer’s disease. Fed. Proc. 45, 2982–2988.
Zakut, H., Matzkel, A., Schejter, E., Avni, A., and Soreq, H. (1985). Polymorphism of acetylcholinesterase in discrete regions of the developing human fetal brain. J. Neurochem. 45, 382–389.
Zhang, Y. W., Thompson, R., Zhang, H., and Xu, H. (2011). APP processing in Alzheimer’s disease. Mol. Brain. 4, 3.
Zimmermann, M., Borroni, B., Cattabeni, F., Padovani, A., and Di Luca, M. (2005). Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol. Dis. 19, 237–242.
Keywords: acetylcholinesterase, β-amyloid, presenilin 1, P-tau, Alzheimer’s disease
Citation: García-Ayllón M-S, Small DH, Avila J and Sáez-Valero J (2011) Revisiting the role of acetylcholinesterase in Alzheimer’s disease: cross-talk with P-tau and β-amyloid. Front. Mol. Neurosci. 4:22. doi: 10.3389/fnmol.2011.00022
Received: 21 June 2011;
Paper pending published: 27 July 2011;
Accepted: 24 August 2011;
Published online: 13 September 2011.
Edited by:
Karl Tsim, The Hong Kong University of Science and Technology, ChinaReviewed by:
Nibaldo C. Inestrosa, Pontifical Catholic University of Chile, ChileYifan Han, Hong Kong Polytechnic University, China
Copyright: © 2011 García-Ayllón, Small, Avila and Sáez-Valero. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Javier Sáez-Valero, Instituto de Neurociencias, Universidad Miguel Hernández-CSIC, E-03550 Sant Joan d’Alacant, Alacant, Spain. e-mail: j.saez@umh.es