Modulating neurotoxicity through CX3CL1/CX3CR1 signaling
 Cristina Limatola
Cristina Limatola Richard M. Ransohoff
Richard M. Ransohoff- 1Department of Physiology and Pharmacology, Istituto Pasteur Fondazione Cenci Bolognetti, Sapienza University of Rome, Rome, Italy
- 2Istituto di Ricovero e Cura a Carattere Scientifico Neuromed, Istituto Neurologico Mediterraneo, Pozzilli, Italy
- 3Neuroinflammation Research Center, Lerner Research Institute and Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, OH, USA
Since the initial cloning of fractalkine/CX3CL1, it was proposed that the only known member of the CX3C or δ subfamily of chemotactic cytokines could play some significant role in the nervous system, due to its high expression on neurons. The pivotal description of the localization of the unique CX3CL1 receptor, CX3CR1, on microglial cells, firmed up by the generation of cx3cr1GFP/GFP mice, opened the road to the hypothesis of some specific key interactions between microglia and neurons mediated by this pair. This expectation has been indeed supported by recent exciting evidence indicating that CX3CL1-mediated microglia-neuron interaction modulates basic physiological activities during development, adulthood and aging, including: synaptic pruning; promoting survival of neurons and neural precursors; modulating synaptic transmission and plasticity; enhancing synapse and network maturation; and facilitating the establishment of neuropathic pain circuits. Beyond playing such fascinating roles in physiological conditions, CX3CL1 signaling has been implicated in different neuropathologies. Early papers demonstrated that the levels of CX3CL1 may be modulated by various toxic stimuli in vitro and that CX3CL1 signaling is positively or negatively regulated in EAE and MS, in HIV infection and LPS challenge, in epilepsy, in brain tumors, and in other neuropathologies. In this review we focus on the experimental evidence of CX3CL1 involvement in neuroprotection and survey the common molecular and cellular mechanisms described in different brain diseases.
The CX3CL1/CX3CR1 axis, together with CD200/CD200R, have mainly been studied in the context of their involvement in halting potentially toxic activated microglial phenotypes (Biber et al., 2007). The phenotypes embodied by the term “microglia activation” have been hotly debated. Recent nomenclatures proposed M1- and M2-like microglial phenotypes, characterized by the production of pro- or anti-inflammatory cytokines and markers, named after the alternative activation states of macrophages (Mantovani et al., 2005). The transition between these two forms, however, is not all or none and several intermediate microglia phenotypes have been described, together with the identification of overlapping features between alternatively activated phenotypes (Ponomarev et al., 2007, 2013; Olah et al., 2012; Crain et al., 2013). Furthermore, it’s now evident that no M1/M2 polarization can be supported, even for peripheral macrophages (Xue et al., 2014).
Given contemporary approaches to genome wide expression profiling, we now face the welcome prospect of devising a robust, meaningful account of microglial reactive phenotypes.
In spite of this complex picture, there is a general consensus on the relatively unique role of the CX3C system in mediating key microglial activities, mainly because of its privileged position at the interface with neurons. Microglia-neuron interaction is dynamic, as revealed by in vivo video microscopy using cx3cr1GFP/+ mice and disclosing that microglia branches continuously survey neuronal surfaces and the cerebral microenvironment in the healthy brain, presumably to sense dysfunctional synapses, damaged neurons, or the presence of potentially dangerous agents (Davalos et al., 2005; Nimmerjahn et al., 2005). Thus, in the adult brain, microglia may exert a sentinel function for neurons and, when neuronal damage occurs, microglia rapidly react to protect or to eliminate neurons, if irreversibly damaged. CX3C signaling is deeply involved in this rescue process, directly modulating different aspects of microglia biology important for neuron protection, like the production of soluble factors directly involved in neuron survival, the modulation of phagocytic activity, but also indirectly affecting other cell types (resident or infiltrating) present in brain parenchyma that, in turn, might influence neuron survival.
CX3CR1 was localized to microglia and CX3CL1 to neurons using in situ hybridization, by Harrison et al. (1998). Availability of mice with a GFP fluorescent reporter for CX3CR1 transcription confirmed the former finding in adult mice (Cardona et al., 2006) and subsequent studies showed that CX3CR1 is characteristic of microglia throughout embryogenesis and during the murine lifespan (Ginhoux et al., 2010; Mizutani et al., 2012; Schulz et al., 2012). Subsequently, Kim et al. (2011) developed a CX3CL1 reporter and showed a similar neuronal distribution of CX3CL1 in all regions of the adult brain. These expression patterns are somewhat modulated but do not fundamentally alter in disease models. Therefore, CX3CL1/CX3CR1 signaling provides insight into microglial–neuronal interactions throughout the lifespan (Table 1) and in an immense variety of pathological conditions. This review summarizes a portion of this research.
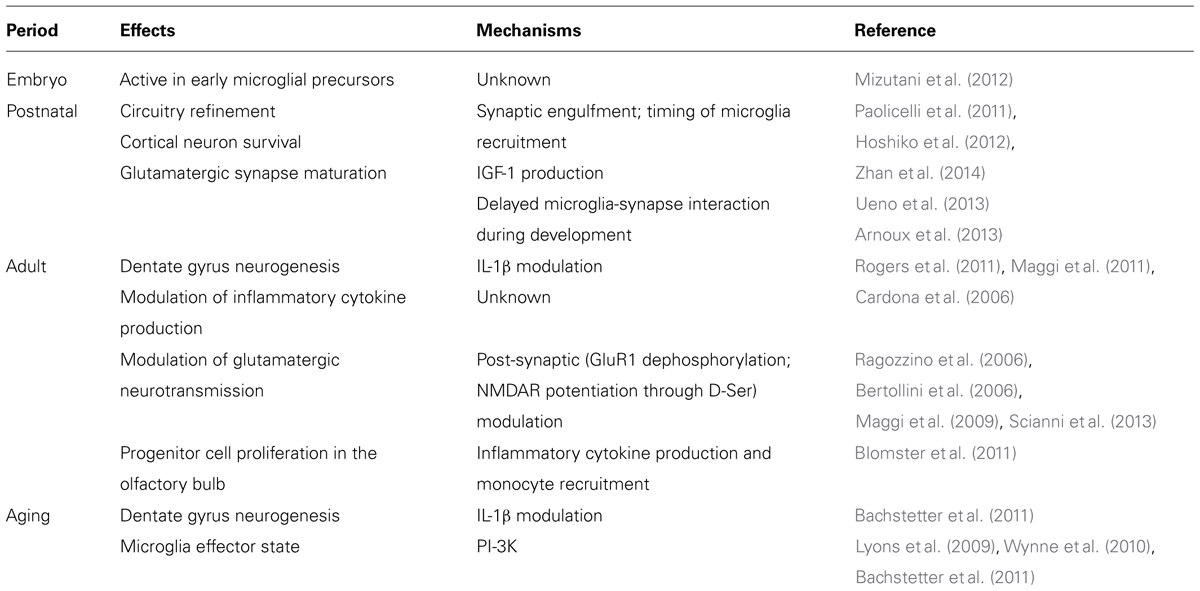
TABLE 1. Documented effects in (ex) vivo of CX3CL1/CX3CR1 signaling in lifespan.
Neurotoxicity Modulation: Cytokine and Growth Factor Production
The CX3CL1/CX3CR1 signaling participates in the control of production and release of several cytokines from microglia. Earlier in vitro studies demonstrated that LPS- and IFNγ-induced release of cytokines such as interleukin-1β (IL-1β), TNFα, 8-isoprostane, NO, and IL-6 in cultured microglia was efficiently blocked by CX3CL1 stimulation (Zujovic et al., 2000, 2001; Mizuno et al., 2003). Since then several papers confirmed and provided further in vivo evidence supporting the hypothesis that, together with other molecules like CD200, CD22 and CD172, CX3CL1 signaling in the brain reduces microglia reactivity to toxic stimuli, maintaining microglia in a modulated state (Biber et al., 2007). This hypothesis can be evaluated even though the status of “microglial activation” is profoundly uncertain [see (Biber et al., 2014) for a recent review].
Modulation of IL-1β Signaling
Interleukin-1β is an inflammatory cytokine playing pivotal roles in local and systemic processes and is a key inducer of peripheral and central immune responses to infection or injury. Inhibition of IL-1β signaling has beneficial effects in a variety of experimental paradigms of acute brain damage. Despite controversial data on its neurotoxic effects in vivo and in vitro, IL-1β is considered a promising clinical target in several neuropathologies (Allan et al., 2005). In paradigms of brain injury where IL-1β increases, CX3CL1 reduces IL-1β levels, correlating with protection from damage.
Increased production of IL-1β by microglial cells isolated from the brain of Cx3cr1-/- mice upon systemic LPS challenge was described by Cardona et al. (2006). Cx3cr1-/- microglial cells from LPS-challenged mice provided a toxic insult when transplanted in the forebrain of wild type mice, resulting in neuronal TUNEL labeling. Interestingly these toxic effects were prevented eliminating IL1R signaling either with IL-1 receptor antagonist a (IL-1Ra) or in IL1-/- mice.
Cardona et al. (2006) also showed enhanced dopaminergic cell loss in Cx3cr1-/- or Cx3cl1-/- mice after peripheral MPTP administration. Morganti et al. (2012) asked two follow-on questions: what was the role of cytokine production? and which CX3CL1 isoform (soluble or membrane-associated) was responsible for the neuroprotective effect? They reported that IL-1β signaling was blunted by soluble AAV-CX3CL1, injected in the substantia nigra pars compacts of MPTP-injected cx3cl1-/- mice, so that less IL-1β and TNFα were produced in the ventral mesencephalon and microglial CD68 expression was reduced. These alterations in cytokine profile induced by soluble AAV-CX3CL1 correlated with reduced death of dopaminergic TH positive neurons and improved motor coordination on accelerating rotarod. A protective effect of soluble CX3CL1 on dopaminergic neurons was demonstrated by the same authors in a rat model of PD, induced by intrastriatal 6-OHDA injection (Pabon et al., 2011).
CX3CL1 decreased in the brain of aged mice possibly due to neuronal loss, and this change is accompanied by an altered effector state of microglia (Lyons et al., 2009; Wynne et al., 2010; Bachstetter et al., 2011). In aged mice, the response to systemic infection is amplified by the microglia effector response: LPS administration evokes an increased IL-1β and a reduced TGFβ expression, in comparison with young adult animals (Wynne et al., 2010).
Aging is reported to decrease neurogenesis in the hippocampal region of the brain (Kuhn et al., 1996). Similarly to aging, deficits in CX3CL1 signaling due to gene deletion or caused by CX3CR1 blocking antibodies reduced the number of progenitor cells in the hippocampal dentate gyrus (Bachstetter et al., 2011; Maggi et al., 2011). CX3CL1 administration reverted the age-related decrease in neurogenesis and chronic blockade of CX3CR1 function reduced neurogenesis and increased IL-1β in young adult mice (Bachstetter et al., 2011). IL-1β was implicated in the reduced survival and proliferation rate of neuronal progenitors in the hippocampal region of CX3CR1-/- mice, since IL-1Ra reverted the observed phenotypes (Bachstetter et al., 2011; Rogers et al., 2011).
The involvement of IL-1 in CX3CL1-induced effects in experimental models of AD is suggested by several lines of evidence. Mouse deleted of cx3cr1 showed a strong increase of microtubule-associated protein tau (MAPT) phosphorylation upon LPS challenge, and humanized MAPT transgenic mice crossed with cx3cr1-/- mice exhibited accelerated behavioral impairment along with increased MAPT hyperphosphorylation and aggregation. These data, together with the observation that microglia-induced MAPT phosphorylation in neurons can be blocked by IL-1Ra and p38 inhibitors further supports the hypothesis that inhibiting production of IL-1β constitutes a central mechanism for CX3CL1 neuroprotective signaling (Bhaskar et al., 2010). Additional possible targets of CX3CL1 signaling in AD have been suggested: Cho et al. (2011) demonstrated that hAPP/cx3cr1-/- mice have exacerbated neuronal and cognitive function defects and increased inflammatory responses in comparison with hAPP/cx3cr1+/+ mice. The lack of cx3cr1 in this AD mouse model (hAPP-J20) did not modify plaque deposition but increased tau phosphorylation, cognitive deficits, and microglia activation, along with enhanced IL-6 and TNF-α levels. In line with these observations, AD autopsy brain sections showed reduced CX3CL1 levels in the cortex and hippocampus regions. Further supporting a potential role for CX3CR1 signaling in AD pathogenesis, Lee et al. (2010) reported that in two different mouse models of AD (APPS1 and R1.40), characterized by different velocity of β-amyloid deposition, crossing with cx3cr1-/- mice reduced the extent of amyloid deposition, the number of dystrophic neurons and of plaque-associated microglia cells. This protected phenotype was accompanied by increased CCL2 and TNF-α and elevated IL-1β expression. By contrast, a potentially toxic effect for CX3CL1 was suggested by Wu et al. (2013) demonstrating that the deficits induced by β-amyloid injection in hippocampal CA1, as well as the increased IL-1β expression and microglia activation, were all reverted by central CX3CR1 suppression by siRNA delivery. A toxic effect for CX3CR1 signaling in AD was also proposed by Fuhrmann et al. (2010) that found a reduced neuronal loss when the 3xTg AD mice were crossed with cx3cr1-/- mice. In summary, CX3CR1/CX3CL1 signaling modifies AD-related pathology quite consistently but a unified concept of the net outcome of the effects has not been conclusively proven.
Alteration of IL-1β levels was reported in cx3cr1-deficient mice upon focal cerebral ischemia. Dénes et al. (2008) demonstrated a reduction in IL-1β and TNF-α production in brains of cx3cr1-/- mice upon induction of cerebral ischemia, together with reduced ischemic volume and less neuronal cell death. Similar experiments in a focal brain ischemia model were performed by Soriano et al. (2002) in cx3cl1 deficient mouse and confirmed lower ischemic volume in the absence of CX3CL1/CX3CR1 signaling. This result was later confirmed in a pMCAO model (Cipriani et al., 2011). In apparent contrast, when CX3CL1 was exogenously administrated to wt mice and followed by pMCAO, a significant reduction of ischemic volume was observed while in cx3cl1-/- mice CX3CL1 administration worsened the effects of ischemia (Cipriani et al., 2011).The mechanisms underlying divergent effects of CX3CL1 administration to wt and Cx3cl1-/- mice remain uncertain. In favor of a protective effect of CX3CL1 in cerebral ischemia, Donohue et al. (2012) reported that stroke patients exhibited a positive correlation between high plasma CX3CL1 levels and a better clinical outcome. Interestingly, Pimentel-Coelho et al. (2013) demonstrated a protective role for CX3CR1 in neonatal hypoxic ischemia in female mice, with a possible gender specific protective effect. The authors demonstrated that hippocampal CX3CL1 was reduced by ischemia and that Cx3cr1-/- mice showed worse learning deficits and hippocampal damage after ischemia (Pimentel-Coelho et al., 2013).
In an in vitro genetic model of amyotrophic lateral sclerosis (ALS), where the mutated SOD1G93A is specifically expressed by astrocytes, Sun et al. (2013) reported that conditioned medium from mesenchymal stem cell (MSC) cultures increased CX3CL1 and reduced TNFα, IL6, and iNOS expression on astrocytes, and mediated a protective effect towards primary motor neurons. In vivo, MSC administration improved the functional outcome of mutated mice. In cultured microglia, the presence of mutated SOD1G93A also increased CX3CR1 levels.
Taken together, these data demonstrate that CX3CL1 attenuates inflammatory cytokine production, strongly modulating the neuroprotective activity of microglia in several brain diseases.
Permissive Effects of Adenosine
Adenosine is a well-established modulator of synaptic transmission and has protective effects in the nervous system, primarily mediated through adenosine type 1 receptor (A1R). Adenosine is released by neurons and glial cells and its level in the brain is regulated essentially by the activity of adenosine kinase (ADK), and 5′ nucleotidase acting on extracellular ATP (Boison et al., 2010).
Upon CX3CL1 treatment of cultured microglia, increased extracellular adenosine release is observed (Lauro et al., 2008, 2010). In in vitro models of excitotoxicity (Lauro et al., 2010) and in vivo models of cerebral ischemia, the neuroprotective activity of CX3CL1 requires A1R activation (Cipriani et al., 2011). This neuroprotective mechanism does not derive from a direct microglia-neuron cross talk, requiring (at least in vitro) the involvement of astrocytes whose A1Rs are stimulated by adenosine released from microglia, increasing astrocyte glutamate transporter (GLT-1) expression and activity (Catalano et al., 2013).
The neuroprotective activity of adenosine has been correlated to its ability to hamper the release of excitatory glutamate at pre-synaptic terminals (Fredholm et al., 1983). In this regard, it is interesting to note that CX3CL1 has a wide spectrum of modulatory activities on glutamatergic neurotransmission, interfering with: (i) pre-synaptic glutamate release, (ii) the amplitude of AMPA currents, thereby altering GluR1 phosphorylation state (post-synaptic modulation), (iii) LTP expression and LTD induction in hippocampal CA1 region, (iv) synaptic NMDAR currents through D-serine release from glia (Meucci et al., 2000; Limatola et al., 2005; Bertollini et al., 2006; Ragozzino et al., 2006; Maggi et al., 2009; Scianni et al., 2013); (v) glutamatergic synapse maturation during development (Paolicelli et al., 2011; Arnoux et al., 2013).
All together these data demonstrate that excitatory neurotransmission, which is deeply implicated in the most common neurotoxic pathways, is affected by CX3CL1/CX3CR1 signaling, with permissive cooperativity of the adenosine system.
Growth Factors Production
Microglia produce a number of growth factors providing trophic support to developing or damaged brain circuits. During brain development, in the first postnatal week, CX3CR1 expression on microglia is important for the survival of cortical neurons of layer V, by regulating the production of insulin-like growth factor-1 (IGF-1; Ueno et al., 2013). In particular, increased death of layer V neurons was observed in the cerebral cortex of cx3cr1GFP/GFP mice, in which microglia produced a significantly reduced amount of the growth factor IGF-1. So during brain development, CX3CR1-mediated IGF-1 secretion from microglia modulates neuronal survival in postnatal cortical layer V.
A specific BDNF polymorphism (Val66Met) is associated with memory deficits and increased vulnerability to anxiety and depressive disorders both in humans and mice. Interestingly, this polymorphism also associates with impaired CX3CL1/CX3CR1 hippocampal signaling, reducing expression of CX3CL1 protein and mRNA in dorsal hippocampus (Wang et al., 2014). Chronic CX3CL1 infusion in the hippocampal region of Val66Met mice recovered memory deficits, restored neurogenesis in the DG and increased Akt phosphorylation levels.
Neurotoxicity Modulation: Microglia Phagocytic Activity
One of the most intriguing functions of microglia in the central nervous system is certainly its activity as the resident phagocyte. In development and in pathological neuroinflammatory conditions, microglial phagocytosis is important for the elimination of dead and damaged neurons, myelin residues and β-amyloid peptides [reviewed in (Sierra et al., 2013)]. During normal brain development, microglial phagocytosis is involved in eliminating supernumerary neurons, but also in synaptic pruning and circuit refinement (Sierra et al., 2010; Tremblay et al., 2010). Among the key factors that emerged as potential modulators of neuron phagocytosis by microglia during development is the complement system, with the proteins C1q and C3 expressed on synapses and microglial CR3/CD11b mediating the elimination of immature and weakly active synapses (Schafer et al., 2012). TGF-β produced by astrocytes signals to retinal ganglion cells for C1q expression (Bialas and Stevens, 2013), thus favoring retinogeniculate refining, and inserting astrocytes in microglia-neuron communication.
Cell damage causes high-grade ATP release (both from damaged cells and through astrocyte exocytosis) regulating microglial process extension (Davalos et al., 2005) and also contributing to inflammasome activation within microglia (Ransohoff and Brown, 2012). Under stress, neurons manifest the phosphatidylserine (PS) “eat me” signal which can be recognized by several microglia receptor/adaptor complexes including Gas6/MerTK and MFGE8/vitronectin receptor (De Simone et al., 2004; Elliott et al., 2009). In ischemic conditions potentially viable neurons reversibly express PS on their surface and are vulnerable to be taken up and destroyed by microglia (Neher et al., 2011). Selective elimination of MFGE8/vitronectin receptor “eat me” signaling strongly improved experimental stroke outcome (Neher et al., 2013).
CX3CR1 signaling also modulates microglia phagocytosis of neurons and their processes in both physiological and pathological conditions. During development, CX3CL1/CX3CR1 signaling contributes to refinement of synaptic elements in varied CNS regions during the postnatal “critical period” (Tremblay et al., 2010; Paolicelli et al., 2011; Hoshiko et al., 2012). This fine-tuning of anatomical connections is important to establish and build an optimal functional circuitry (Zhan et al., 2014).
In Alzheimer’s disease (AD), CX3CL1/CX3CR1 blockade modulates microglia phagocytic activity in a way that markedly attenuates amyloid deposition (Lee et al., 2010; Liu et al., 2010). In particular, CX3CR1 deficient microglia show increased phagocytic activity when crossed with different AD mouse models CRND8 (Liu et al., 2010) or APPPS1 and R1.40 (Lee et al., 2010), resulting in attenuated Aβ deposition. Based on results that look beyond amyloid deposition in various models, the overall outcome of eliminating CX3CR1 signaling remains in doubt (Bhaskar et al., 2010; Fuhrmann et al., 2010; Cho et al., 2011).
Neurons damaged by toxic insults, like glutamate, release more CX3CL1 that increases MFG-E8 expression on microglia (Leonardi-Essmann et al., 2005; Fuller and Van Eldik, 2008; Noda et al., 2011). As noted above, MFG-E8 is a PS receptor that recognize PS on injured neurons and acts as an adaptor recognized by microglial vitronectin receptor, inducing damaged-cell uptake. CX3CL1 up-regulation of MFG-E8 expression was associated to increased heme oxygenase 1 (HO-1) expression through the MAPKs ERK and JNK and the nuclear factor erythroid 2 related factor (NFE2LE). Lastres-Becker et al. (2014) reported that CX3CL1 reduced Tau-induced microgliosis via up-regulating transcription factor NRF2/NFE2LE, along with increased HO-1 expression. Since the increased expression of HO-1 is reported to mediate anti inflammatory activities (Chora et al., 2007), this pathway could be involved in the reported neuroprotective effects of CX3CL1 in different Tau pathology models (Nash et al., 2013).
These data indicate that the clearance and phagocytic activity of microglia can be modulated by CX3CL1 signaling through varied mechanisms, and that the final outcome in term of neuroprotection or neurotoxicity might result from the presence of co-stimulatory signals and, consequently, from the effector programs of microglia.
Neurotoxicity Modulation: Effects on Neural Precursors
In vitro, CX3CL1 promoted survival of neural precursor cells upon growth factor withdrawal from the culture medium (Krathwohl and Kaiser, 2004). Further, cx3cr1-/- mice have reduced neurogenesis in the hippocampal DG region (Bachstetter et al., 2011; Maggi et al., 2011) but this reduction was accompanied by contrasting effects on memory deficits and synaptic plasticity: Maggi et al., (Maggi et al., 2011) reported that cx3cr1-/- mice had increased hippocampal LTP and performed better in the Morris water maze test, while, in the same mouse strain, Rogers et al. (2011) described reduced hippocampal LTP and deficits in the Morris water maze. At least in part, these conflicting data reflect the lack of simple relations between DG neurogenesis, plasticity processes in the hippocampal CA1 region and learning behavior.
More recently, Vukovic et al. (2012) demonstrated that CX3CR1 deficiency impairs neurogenesis both in young and aged mice and that voluntary physical exercise increased brain CX3CL1 levels and DG neurogenesis, both effects being abolished by intrahippocampal CX3CR1 antibody injection.
In the olfactory bulb, CX3CR1 deletion increased olfactory sensory neuron death upon bulbectomy and cx3cr1-/- mice showed reduced proliferation of intraepithelial stem progenitor cells, increased macrophage recruitment in the lesioned epithelium and increased TNF-α and IL-6 levels (Blomster et al., 2011).
All together these data suggest that neuroprotective effects of CX3CL1 might arise partly from an increased proliferation or survival of progenitor elements in selected brain regions where neurogenesis continues throughout adult life.
Neurotoxicity Modulation: Indirect Effects
Modulation of Astrocyte Activity
In the CNS, the unique expression of CX3CR1 on microglia clearly suggests that the neuroprotective effects of CX3CL1 depend primarily on the direct communication between microglia and neurons. Recent evidence suggested that CX3CL1/CX3CR1 signaling in neuroprotection again excitotoxic insult might not be restricted to a direct microglia-neuron communication but also involves indirectly modulation of astrocyte activity. Catalano et al. (2013), in fact, demonstrated that CX3CL1, acting on microglia, induced the production and release of soluble factors that exerted their effects on astrocytes, inducing the functional up regulation and the increased expression of the excitatory amino acid transporter GLT-1. As already reported in in vitro and in vivo systems (Lauro et al., 2010; Cipriani et al., 2011), this cross-talk requires the “permissive” presence of adenosine, specifically acting on astrocyte A1R (Catalano et al., 2013). These data demonstrated for the first time a role for astrocytes in mediating the neuroprotection induced by CX3CL1/CX3CR1 signaling between microglia and neurons.
Effects Through Leukocyte Recruitment
Neuroprotective effects of CX3CL1 could also arise from hematogenous leukocytes that enter the brain upon specific, injury-induced challenge.
In mouse experimental allergic encephalomyelitis (EAE) Huang et al. (2006) demonstrated that CX3CR1 deficient mice had an exacerbated phenotype, with severe spastic paralysis and hemorrhagic inflammation in the CNS, resulting in higher mortality. The increased toxicity resulted from an altered control of NK cells entry into the brain of EAE-affected animals. These findings were extended by Garcia et al. (2013), who showed that CX3CR1 deficiency limited to bone marrow cells of mice with EAE resulted in more severe pathology, with increased demyelination, axonal damage and reduced calbindin positive neurons in the cerebellum. These effects correlated with increased CD115+, Ly6ClowCD11c+ dendritic cell infiltration in the CNS and increased IL-17 and IFNγ expression in the cerebellum, forebrain and spinal cord.
Mills et al. (2008) reported that in CD73-/- mice, where the ectonucleotidase responsible for extracellular adenosine accumulation was absent, EAE induction produced a milder phenotype. More recently, the same group (Mills et al., 2012) reported that extracellular adenosine increased CX3CL1 expression and that CX3CL1 promoted lymphocyte entry into the brain of EAE mice. Therefore, in CD73 deficient mice, extracellular adenosine cannot be produced, CX3CL1 is not upregulated and a reduced amount of lymphocytes enter cerebral parenchyma, resulting in a protected phenotype (Mills et al., 2012).
In focal cerebral ischemia, Dénes et al. (2008) reported a reduced leukocyte (CD45+ cells) infiltration in the brain of cx3cr1-/- mice and a consistently reduced damaging of the blood brain barrier. This correlated with a protected phenotype, with reduced ischemic volume and lesser numbers of apoptotic cells and a better performance in the behavioral test of adhesive tape removal.
All together these data highlight that CX3CL1/CX3CR1 signaling modulates the entry of hematogenous leukocytes, with beneficial or deleterious consequences contingent on context.
Conclusion
This review highlights the manifold effects of altering CX3CR1/CX3CL1 signaling, which can be observed throughout the lifespan from early embryogenesis through diseases of the aging brain. For the most part, CX3CR1/CX3CL1 can be viewed as a fulcrum with which to modify microglial physiology to probe into the panoply of functions of these versatile and essential cells. Adding to complexity but also lending heuristic value, the effects of modifying CX3CR1/CX3CL1 pathways are extremely context dependent. Nevertheless in each case insights about upstream and downstream signaling from CX3CR1/CX3CL1 have been informative and may point to new therapeutic directions for brain disease. Finally, it is now clear that macroglia (such as astrocytes) and hematogenous leukocytes also contribute to the varied physiology of the CX3CR1/CX3CL1 signaling system.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the funding for research in their laboratories: Cristina Limatola: AIRC IG 2012; Richard M. Ransohoff: NIH NS32151; National MS Society RG4552; Charles A. Dana Foundation; Williams Foundation Fund for MS Research.
References
Allan, S. M., Tyrrell, P. J., and Rothwell, N. J. (2005). Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 5, 629–640. doi: 10.1038/nri1664
Arnoux, I., Hoshiko, M., Mandavy, L., Avignone, E., Yamamoto, N., and Audinat, E. (2013). Adaptive phenotype of microglial cells during the normal postnatal development of the somatosensory “Barrel” cortex. Glia 61, 1582–1594. doi: 10.1002/glia.22503
Bachstetter, A. D., Morganti, J. M., Jernberg, J., Schlunk, A., Mitchell, S. H., Brewster, K. W.,et al. (2011). Fractalkine and CX3CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 32, 2030–2044. doi: 10.1016/j.neurobiolaging.2009.11.022
Bertollini, C., Ragozzino, D., Gross, C., Limatola, C., and Eusebi F. (2006). Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacol. 51, 816–821. doi: 10.1016/j.neuropharm.2006.05.027
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi: 10.1016/j.neuron.2010.08.023
Bialas, A. R., and Stevens, B. (2013). TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 16, 1773–1782. doi: 10.1038/nn.3560
Biber, K., Neumann, H., Inoue, K., and Boddeke, H. W. (2007). Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 30, 596–602. doi: 10.1016/j.tins.2007.08.007
Biber, K., Owens, T., and Boddeke, E. (2014). What is microglia neurotoxicity (Not)? Glia 62, 841–854. doi: 10.1002/glia.22654
Blomster, L. V., Vukovic, J., Hendrickx, D. A., Jung, S., Harvey, A. R., Filgueira, L.,et al. (2011). CX3CR1 deficiency exacerbates neuronal loss and impairs early regenerative responses in the target-ablated olfactory epithelium. Mol. Cell. Neurosci. 48, 236–245. doi: 10.1016/j.mcn.2011.08.004
Boison, D., Chen, J. F., and Fredholm, B. B. (2010). Adenosine signaling and function in glial cells. Cell Death Differ. 17, 1071–1082. doi: 10.1038/cdd.2009.131
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M.,et al. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924. doi: 10.1038/nn1715
Catalano, M., Lauro, C., Cipriani, R., Chece, G., Ponzetta, A., Di Angelantonio, S.,et al. (2013). CX3CL1 protects neurons against excitotoxicity enhancing GLT-1 activity on astrocytes. J. Neuroimmunol. 263, 75–82. doi: 10.1016/j.jneuroim.2013.07.020
Cho, S. H., Sun, B., Zhou, Y., Kauppinen, T. M., Halabisky, B., Wes, P.,et al. (2011). CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 286, 32713–32722. doi: 10.1074/jbc.M111.254268
Chora, A. A., Fontoura, P., Cunha, A., Pais, T. F., Cardoso, S., Ho, P. P.,et al. (2007). Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J. Clin. Invest. 117, 438–447. doi: 10.1172/JCI28844
Cipriani, R., Villa, P., Chece, G., Lauro, C., Paladini, A., Micotti, E.,et al. (2011). CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J. Neurosci. 31, 16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011
Crain, J. M., Nikodemova, M., and Watters, J. J. (2013). Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J. Neurosci. Res. 91, 1143–1151. doi: 10.1002/jnr.23242
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S.,et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
De Simone, R., Ajmone-Cat, M. A., and Minghetti, L. (2004). Atypical antiinflammatory activation of microglia induced by apoptotic neurons: possible role of phosphatidylserine receptor interaction. Mol. Neurobiol. 29, 197–212. doi: 10.1385/MN:29:2:197
Dénes, A., Ferenczi, S., Halász, J., Környei, Z., and Kovács, K. J. (2008). Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 28, 1707–1721. doi: 10.1038/jcbfm.2008.64
Donohue, M. M., Cain, K., Zierath, D., Shibata, D., Tanzi, P. M., and Becker, K. J. (2012). Higher plasma fractalkine is associated with better 6-month outcome from ischemic stroke. Stroke 43, 2300–2306. doi: 10.1161/STROKEAHA.112.657411
Elliott, M. R., Chekeni, F. B., Trampont, P. C., Lazarowski, E. R., Kadl, A., Walk, S. F.,et al. (2009). Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286. doi: 10.1038/nature08296
Fredholm, B. B., Jonzon, B., and Lindgren, E. (1983). Inhibition of noradrenaline release from hippocampal slices by a stable adenosine analogue. Acta Physiol. Scand Suppl. 515, 7–10.
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G.,et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Fuller, A. D., and Van Eldik, L. J. (2008). MFG-E8 regulates microglial phagocytosis of apoptotic neurons. J. Neuroimmune Pharmacol. 3, 246–256. doi: 10.1007/s11481-008-9118-2
Garcia, J. A., Pino, P. A., Mizutani, M., Cardona, S. M., Charo, I. F., Ransohoff, R. M.,et al. (2013). Regulation of adaptive immunity by the fractalkine receptor during autoimmune inflammation. J. Immunol. 191, 1063–1072. doi: 10.4049/jimmunol.1300040
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S.,et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., McNamara, R. K.,et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U.S.A. 95, 10896–10901. doi: 10.1073/pnas.95.18.10896
Hoshiko, M., Arnoux, I., Avignone, E., Yamamoto, N., and Audinat, E. (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 32, 15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012
Huang, D., Shi, F. D., Jung, S., Pien, G. C., Wang, J., Salazar-Mather, T. P.,et al. (2006). The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 20, 896–905. doi:10.1096/fj.05-5465com
Kim, K. W., Vallon-Eberhard, A., Zigmond, E., Farache, J., Shezen, E., Shakhar, G.,et al. (2011). In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 118:e156–e167. doi: 10.1182/blood-2011-04-34894619
Krathwohl, M. D., and Kaiser, J. L. (2004). Chemokines promote quiescence and survival of human neural progenitor cells. Stem Cells 22, 109–118. doi: 10.1634/stemcells.22-1-109
Kuhn, H. G., Dickinson-Anson, H., and Gage, F. H. (1996). Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16, 2027–2033.
Lastres-Becker, I., Innamorato, N. G., Jaworski, T., Rábano, A., Kügler, S., Van Leuven, F.,et al. (2014). Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 137, 78–91. doi: 10.1093/brain/awt323
Lauro, C., Cipriani, R., Catalano, M., Trettel, F., Chece, G., Brusadin, V.,et al. (2010). Adenosine A1 receptors and microglial cells mediate CX3CL1-induced protection of hippocampal neurons against Glu-induced death. Neuropsychopharmacol. 35, 1550–1559. doi: 10.1038/npp.2010.26
Lauro, C., Di Angelantonio, S., Cipriani, R., Sobrero, F., Antonilli, L., Brusadin, V.,et al. (2008). Activity of adenosine receptors type 1 is required for CX3CL1-mediated neuroprotection and neuromodulation in hippocampal neurons. J. Immunol. 180, 7590–7596. doi: 10.4049/jimmunol.180.11.7590
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M.,et al. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 177, 2549–2562. doi: 10.2353/ajpath.2010.100265
Leonardi-Essmann, F., Emig, M., Kitamura, Y., Spanagel, R., and Gebicke-Haerter, P. J. (2005). Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. J. Neuroimmunol. 160, 92–101. doi: 10.1016/j.jneuroim.2004.11.012
Limatola, C., Lauro, C., Catalano, M., Ciotti, M. T., Bertollini, C., Di Angelantonio, S.,et al. (2005). Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J. Neuroimmunol. 166, 19–28. doi: 10.1016/j.jneuroim.2005.03.023
Liu, Z., Condello, C., Schain, A., Harb, R., and Grutzendler, J. (2010). CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J. Neurosci. 30, 17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010
Lyons, A., Lynch, A. M., Downer, E. J., Hanley, R., O’Sullivan, J. B., Smith, A.,et al. (2009). Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem. 110, 1547–5156. doi: 10.1111/j.1471-4159.2009.06253.x
Maggi, L., Scianni, M., Branchi, I., D’Andrea, I., Lauro, C., and Limatola, C. (2011). CX(3)CR1 deficiency alters hippocampal-dependent plasticity phenomena blunting the effects of enriched environment. Front Cell Neurosci. 5:22. doi: 10.3389/fncel.2011.00022
Maggi, L., Trettel, F., Scianni, M., Bertollini, C., Eusebi, F., Fredholm, B. B.,et al. (2009). LTP impairment by fractalkine/CX3CL1 in mouse hippocampus is mediated through the activity of adenosine receptor type 3 (A3R). J. Neuroimmunol. 215, 36–42. doi: 10.1016/j.jneuroim.2009.07.016
Mantovani, A., Sica, A., and Locati, M. (2005). Macrophage polarization comes of age. Immunity 23, 344–346. doi: 10.1016/j.immuni.2005.10.001
Meucci, O., Fatatis, A., Simen, A. A., and Miller, R. J. (2000). Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. U.S.A. 97, 8075–8080. doi: 10.1073/pnas.090017497
Mills, J. H., Alabanza, L. M., Mahamed, D. A., and Bynoe, M. S. (2012). Extracellular adenosine signaling induces CX3CL1 expression in the brain to promote experimental autoimmune encephalomyelitis. J. Neuroinflammation 9:193. doi: 10.1186/1742-2094-9-193
Mills, J. H., Thompson, L. F., Mueller, C., Waickman, A. T., Jalkanen, S., Niemela, J.,et al. (2008). CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 105, 9325–9330. doi: 10.1073/pnas.0711175105
Mizuno, T., Kawanokuchi, J., Numata, K., and Suzumura, A. (2003). Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 979, 65–70. doi: 10.1016/S0006-8993(03)02867-1
Mizutani, M., Pino, P. A., Saederup, N., Charo, I. F., Ransohoff, R. M., and Cardona, A. E. (2012). The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J. Immunol. 188, 29–36. doi: 10.4049/jimmunol.1100421
Morganti, J. M., Nash, K. R., Grimmig, B. A., Ranjit, S., Small, B., Bickford, P. C.,et al. (2012). The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci. 32, 14592–14601. doi: 10.1523/JNEUROSCI.0539-12.2012
Nash, K. R., Lee, D. C., Hunt, J. B. Jr., Morganti, J. M., Selenica, M. L., Moran, P.,et al. (2013). Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 34, 1540–1548. doi: 10.1016/j.neurobiolaging.2012.12.011
Neher, J. J., Emmrich, J. V., Fricker, M., Mander, P. K., Théry, C., and Brown, G. C. (2013). Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. U.S.A. 110, E4098–E4107. doi: 10.1073/pnas.1308679110
Neher, J. J., Neniskyte, U., Zhao, J. W., Bal-Price, A., Tolkovsky, A. M., and Brown, G. C. (2011). Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 186, 4973–4983. doi: 10.4049/jimmunol.1003600
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Noda, M., Doi, Y., Liang, J., Kawanokuchi, J., Sonobe, Y., Takeuchi, H.,et al. (2011). Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 286, 2308–2319. doi: 10.1074/jbc.M110.169839
Olah, M., Amor, S., Brouwer, N., Vinet, J., Eggen, B., Biber, K.,et al. (2012). Identification of a microglia phenotype supportive of remyelination. Glia 60, 306–321. doi: 10.1002/glia.21266
Pabon, M. M., Bachstetter, A. D., Hudson, C. E., Gemma, C., and Bickford, P. C. (2011). CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J. Neuroinflammation 8:9. doi: 10.1186/1742-2094-8-9
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P.,et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Pimentel-Coelho, P. M., Michaud, J. P., and Rivest, S. (2013). Evidence for a gender-specific protective role of innate immune receptors in a model of perinatal brain injury. J. Neurosci. 33, 11556–11572. doi: 10.1523/JNEUROSCI.0535-13.2013
Ponomarev, E. D., Maresz, K., Tan, Y., and Dittel, B. N. (2007). CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 27, 10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007
Ponomarev, E. D., Veremeyko, T., and Weiner, H. L. (2013). MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 61, 91–103. doi: 10.1002/glia.22363
Ragozzino, D., Di Angelantonio, S., Trettel, F., Bertollini, C., Maggi, L., Gross, C.,et al. (2006). Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. J. Neurosci. 26, 10488–10498. doi: 10.1523/JNEUROSCI.3192-06.2006
Ransohoff, R. M., and Brown, M. A. (2012). Innate immunity in the central nervous system. J. Clin. Invest. 122, 1164–1171. doi: 10.1172/JCI58644
Rogers, J. T., Morganti, J. M., Bachstetter, A. D., Hudson, C. E., Peters, M. M., Grimmig, B. A.,et al. (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R.,et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schulz, C., Gomez Perdiguero, E., Chorro, L., Szabo-Rogers, H., Cagnard, N., Kierdorf, K.,et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. doi: 10.1126/science.1219179
Scianni, M., Antonilli, L., Chece, G., Cristalli, G., Di Castro, M. A., Limatola, C.,et al. (2013). Fractalkine (CX3CL1) enhances hippocampal N-methyl-D-aspartate receptor (NMDAR) function via D-serine and adenosine receptor type A2 (A2AR) activity. J. Neuroinflammation 10:108. doi: 10.1186/1742-2094-10–108
Sierra, A., Abiega, O., Shahraz, A., and Neumann, H. (2013). Janus-faced microglia: beneficial and detrimental consequences of microglia phagocytosis. Front. Cell. Neurosci. 7:1–22. doi: 10.3389/fncel.2013.00006
Sierra, A., Encinas, J. M., Deudero, J. J., Chancey, J. H., Enikolopov, G., Overstreet-Wadiche, L. S.,et al. (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495. doi: 10.1016/j.stem.2010.08.014
Soriano, S. G., Amaravadi, L. S., Wang, Y. F., Zhou, H., Yu, G. X., Tonra, J. R.,et al. (2002). Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J. Neuroimmunol. 125, 59–65. doi: 10.1016/S0165-5728(02)00033-4
Sun, H., Bénardais, K., Stanslowsky, N., Thau-Habermann, N., Hensel, N., Huang, D.,et al. (2013). Therapeutic potential of mesenchymal stromal cells and MSC conditioned medium in Amyotrophic Lateral Sclerosis (ALS)–in vitro evidence from primary motor neuron cultures, NSC-34 cells, astrocytes and microglia. PLoS ONE 8:e72926. doi: 10.1371/journal.pone.0072926
Tremblay, M.È., Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Ueno, M., Fujita, Y., Tanaka, T., Nakamura, Y., Kikuta, J., Ishii, M.,et al. (2013). Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 16, 543–551. doi: 10.1038/nn.3358
Vukovic, J., Colditz, M. J., Blackmore, D. G., Ruitenberg, M. J., and Bartlett, P. F. (2012). Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J. Neurosci. 32, 6435–6443. doi: 10.1523/JNEUROSCI.5925-11.2012
Wang, D. D., Tian, T., Dong, Q., Xu, X. F., Yu, H., Wang, Y.,et al. (2014). Transcriptome profiling analysis of the mechanisms underlying the BDNF Val66Met polymorphism induced dysfunctions of the central nervous system. Hippocampus 24, 65–78. doi: 10.1002/hipo.22204
Wu, J., Bie, B., Yang, H., Xu, J. J., Brown, D. L., and Naguib, M. (2013). Suppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiency. Neurobiol. Aging 34, 2843–2852. doi: 10.1016/j.neurobiolaging.2013.06.003
Wynne, A. M., Henry, C. J., Huang, Y., Cleland, A., and Godbout, J. P. (2010). Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 24, 1190–1201. doi: 10.1016/j.bbi.2010.05.011
Xue, J., Schmidt, S. V., Sander, J., Draffehn, A., Krebs, W., Quester, I.,et al. (2014). Trancriptome- based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. doi: 10.1016/j.immuni.2014.01.006
Zhan, Y., Paolicelli, R. C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F.,et al. (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406. doi: 10.1038/nn.3641
Zujovic, V.,Benavides, J., Vigé, X., Carter, C., and Taupin, V. (2000). Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia 29, 305–315. doi: 10.1002/(SICI)1098-1136(20000215)29:4<305::AID-GLIA2>3.0.CO;2-V
Zujovic, V., Schussler, N., Jourdain, D., Duverger, D., and Taupin, V. (2001). In vivo neutralization of endogenous brain fractalkine increases hippocampal TNFalpha and 8-isoprostane production induced by intracerebroventricular injection of LPS. J. Neuroimmunol. 115, 135–143. doi: 10.1016/S0165-5728(01)00259-4
Keywords: CX3CL1, CX3CR1, microglia, neurotoxicity, signaling
Citation: Limatola C and Ransohoff RM (2014) Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front. Cell. Neurosci. 8:229. doi: 10.3389/fncel.2014.00229
Received: 07 May 2014; Accepted: 23 July 2014;
Published online: 08 August 2014.
Edited by:
Shawn Hayley, Carleton University, CanadaReviewed by:
Carmelina Gemma, University of Washington, USAJeffrey Keith Harrison, University of Florida, USA
Copyright © 2014 Limatola and Ransohoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Limatola, Department of Physiology and Pharmacology, Sapienza University, Piazzale Aldo Moro 5, Rome 00185, Italy e-mail: cristina.limatola@uniroma1.it