
Abstract
We found that leptin receptors were expressed in hypothalamic astrocytes and that their conditional deletion led to altered glial morphology and synaptic inputs onto hypothalamic neurons involved in feeding control. Leptin-regulated feeding was diminished, whereas feeding after fasting or ghrelin administration was elevated in mice with astrocyte-specific leptin receptor deficiency. These data reveal an active role of glial cells in hypothalamic synaptic remodeling and control of feeding by leptin.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Accession codes
Change history
10 June 2014
In the PDF version of the supplementary information initially published, Supplementary Figures 1a,b and 5a and the corresponding legends were missing. In the HTML version, the legends were present but the figure panels were missing. The errors have been corrected in the HTML and PDF versions of the supplementary information as of 10 June 2014.
References
Bélanger, M., Allaman, I. & Magistretti, P.J. Cell Metab. 14, 724–738 (2011).
Allaman, I., Belanger, M. & Magistretti, P.J. Trends Neurosci. 34, 76–87 (2011).
West, M.J. & Gundersen, H.J. J. Comp. Neurol. 296, 1–22 (1990).
Diano, S., Naftolin, F. & Horvath, T.L. J. Neuroendocrinol. 10, 239–247 (1998).
Horvath, T.L. et al. Proc. Natl. Acad. Sci. USA 107, 14875–14880 (2010).
Fuente-Martín, E. et al. J. Clin. Invest. 122, 3900–3913 (2012).
Sholl, D.A. J. Anat. 93, 143–158 (1959).
Hsuchou, H., Pan, W., Barnes, M.J. & Kastin, A.J. Peptides 30, 2275–2280 (2009).
Hsuchou, H. et al. Brain 132, 889–902 (2009).
Ganat, Y.M. et al. J. Neurosci. 26, 8609–8621 (2006).
García-Cáceres, C. et al. Endocrinology 152, 1809–1818 (2011).
Gao, Q. & Horvath, T.L. Annu. Rev. Neurosci. 30, 367–398 (2007).
Cowley, M.A. et al. Nature 411, 480–484 (2001).
Vong, L. et al. Neuron 71, 142–154 (2011).
Cowley, M.A. et al. Neuron 37, 649–661 (2003).
Andrews, Z.B. et al. Nature 454, 846–851 (2008).
Yang, Y., Atasoy, D., Su, H.H. & Sternson, S.M. Cell 146, 992–1003 (2011).
McMinn, J.E. et al. Am. J. Physiol. Endocrinol. Metab. 289, E403–E411 (2005).
Patterson, C.M., Leshan, R.L., Jones, J.C. & Myers, M.G. Jr. Brain Res. 1378, 18–28 (2011).
Diano, S. et al. Nat. Neurosci. 9, 381–388 (2006).
Caron, E., Sachot, C., Prevot, V. & Bouret, S.G. J. Comp. Neurol. 518, 459–476 (2010).
Del Cerro, S., Garcia-Estrada, J. & Garcia-Segura, L.M. Glia 14, 65–71 (1995).
Jeong, J.K., Chen, Z., Tremere, L.A. & Pinaud, R. J. Vis. Exp. 42, 2102 (2010).
Sanz, E. et al. Proc. Natl. Acad. Sci. USA 106, 13939–13944 (2009).
Heiman, M. et al. Cell 135, 738–748 (2008).
Doyle, J.P. et al. Cell 135, 749–762 (2008).
Dougherty, J.D., Schmidt, E.F., Nakajima, M. & Heintz, N. Nucleic Acids Res. 38, 4218–4230 (2010).
Dietrich, M.O. et al. Nat. Neurosci. 15, 1108–1110 (2012).
Acknowledgements
This work was supported by the US National Institutes of Health (DP1 DK098058, R01AG040236, P01NS062686 and R01 DK097566), the American Diabetes Association, the Helmholtz Society (ICEMED) and the Klarmann Family Foundation.
Author information
Authors and Affiliations
Contributions
J.G.K., M.O.D. and T.L.H. designed the study. J.G.K., M.H.T. and T.L.H. interpreted the results. J.G.K. and S.J. performed the experiments and analyzed the data. M.K. and K.S.-B. contributed to Figures 1h,j and 2a–c. J.K.J. and S.D. contributed to Figure 1c. S.S. and Z.-W.L. contributed to Figure 2d–g and Supplementary Figure 6. M.R.Z., N.S. and F.M.V. contributed to Supplementary Figures 1 and 4a. P.A.-A., J.C. and J.A. contributed to Supplementary Figure 2b. Y.G., C.G.-C. and C.-X.Y. contributed to the generation of the animal model. J.G.K. and T.L.H. wrote the paper with input from the other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
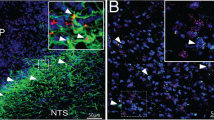
Supplementary Figure 1 Presence of leptin receptors in hypothalamic astrocytes.
(a) Double fluorescence labeling of the astrocyte marker GFAP (red) and leptin receptors (leptin receptor-driven expression of EGFP, green) shows co-localization of GFAP-immunolabeling with EGFP-tagged leptin receptor-containing profiles (white arrows) in the arcuate nucleus (Arc). Scale bar = 100 μm. (b) The truncated leptin receptor (exon 17) allele was confirmed by in situ hybridization combined with immunohistochemistry. Red fluorescence indicates GFAP-positive astrocyte and green fluorescence indicates POMC neurons. White dots indicate mRNA signals of the leptin receptor containing exon 17. White arrows indicate cells expressing mRNA of leptin receptors. White arrowheads indicate leptin receptor-negative cells. Scale bar = 30 μm. (c) To further confirm astrocyte-specific expression of leptin receptors, transcript of leptin receptor b (LepRb) was amplified from RNA bound to the ribosomes selectively in astrocytes isolated from AldH-EGFP-L10a mice and in AgRP neurons isolated from AgRP-cre:floxed Rpl22HA mice. To check purification and contamination of the samples, we analyzed enrichment of AgRP (marker for AgRP purification), NeuN (Marker for neuronal cell contamination) and s100β (marker for astrocyte contamination) compared to that of input samples. Purple bar graphs represent enrichment of LepRb in the AgRP neurons or astrocytes of the hypothalamus compared to that of input controls.
Supplementary Figure 2 Strategy of transgenic mice with selective impairment of leptin receptor signaling in astrocytes.
(a) Schematic drawing of floxed leptin receptor construct, which was used to generate a mouse line allowing cell and time-specific knockout of leptin receptors in astrocytes. (b) RT-PCR shows a deletion of exon 17 of the leptin receptor in primary astrocytes from GFAP-LepR–/– mice. Representative images were selected from at least 3 time repeated experiments.
Supplementary Figure 3 Verification of astrocyte-specific Cre-recombination.
(a) Schematic drawing of human GFAP-driven Cre promoter construct under control of estrogen receptors. (b) GFAP-CreERT2 mice were crossed with tdTomato-loxP reporter mice to confirm successful Cre-mediated recombination in GFAP-positive cells. (c) Ai14 tdTomato mice express red fluorescence in GFAP-positive cells following cre-mediated recombination. tdTomato fluorescences were not co-localized with signals of Iba-1, a biomarker for microglia or NeuN, a biomarker for neuron. White arrows indicate double-labeled cells. Representative images were selected from at least 3 time repeated experiments. Scale bar = 50 μm.
Supplementary Figure 4 Effect of astrocyte-specific deletion of leptin receptors on morphology of astrocytes in the hippocampal CA3.
(a) To confirm expression of leptin receptor in hippocampal astrocytes, we performed ribosome profiling to isolated RNA bound to the ribosomes selectively from hippocampal astrocytes and analyzed enrichment of LepRb. Purple bar graph represents enrichment of LepRb in astrocytes of the hippocampus compared to that of input control. (b) Representative image of GFAP-immunolabeling in the hippocampal CA3 of GFAP-LepR+/+ (+/+) or GFAP-LepR–/– (–/–) mice. (c) The number of GFAP-positive cells (n=5 slices for GFAP-LepR+/+; n=4 slices for GFAP-LepR–/–, p=0.7150, t(7)=0.3803) and (d) length (n=8 cells for GFAP-LepR+/+; n=8 cells for GFAP-LepR–/–, p>0.9999, t(14)=0.0) and (e) number (n=8 cells for GFAP-LepR+/+; n=8 cells for GFAP-LepR–/–, p=0.456, t(14)=0.7667) of astrocyte primary projections did not differ between GFAP-LepR+/+ and GFAP-LepR–/–mice. Results are means ± the s.e.m. P values for unpaired comparisons were analyzed by two-tailed Student's t-test.
Supplementary Figure 5 Leptin receptor signaling in astrocytes affects contact formation between astrocytes and melanocortin cells.
(a) Representative electron micrograph showing astrocyte coverage (green pseudo-color and blue arrows) onto POMC-labeled cells. POMC cells of GFAP-LepR–/– mice had less coverage of their perikaryal membranes by astrocytic processes compared to controls. Scale bar = 1 μm. Coronal sections labeled with GFP in AgRP or POMC cells were incubated with GFAP antibody. (b, d) Representative pictures of double-labeled AgRP or POMC-GFP and GFAP-positive cells in the Arc of GFAP-LepR+/+ or GFAP-LepR–/–mice. Percentage of (c) POMC (n=7 slices for GFAP-LepR+/+; n=6 slices for GFAP-LepR–/–, p<0.0001, t(11)=8.226) or (e) AgRP cells (n=8 slices for GFAP-LepR+/+; n=8 slices for GFAP-LepR–/–, p=0.0004, t(14)=4.672) contacted with GFAP-fiber signals was reduced in GFAP-LepR–/– mice. White arrows indicate cells interacted with astrocyte fibers. ***, p<0.001 versus GFAP-LepR+/+ mice. Results are means ± the s.e.m. P values for unpaired comparisons were analyzed by two-tailed Student's t-test. Scale bar = 100 μm.
Supplementary Figure 6 The probability distribution and average peak amplitude of miniature postsynaptic currents on AgRP or POMC neurons.
(a, b) Cummulative probability distribution and average peak amplitude (inserts) of mIPSC or mEPSC onto AgRP neurons (Fig. 6a: n=9 cells for GFAP-LepR+/+; n=9 cells for GFAP-LepR–/–, p=0.7715, t(798)=0.2906; Fig. 6b: n=9 cells for GFAP-LepR+/+; n=9 cells for GFAP-LepR–/–, p=0.2788, t(798)=1.084). No-differences were observed between control and GFAP-LepR–/– mice. (c) GFAP-LepR–/– mice showed a significantly increase in probability distribution and average peak amplitude (inserts) of mIPSC onto POMC neurons (n=9 cells for GFAP-LepR+/+; n=9 cells for GFAP-LepR–/–, p<0.0001, t(896)=7.604). (d) GFAP-LepR–/– mice revealed an increase in probability distribution of mEPSC amplitude onto POMC neurons (n=23 cells for GFAP-LepR+/+; n=25 cells for GFAP-LepR–/–, p<0.0001, t(3998)=5.255). **, p<0.01; ***, p<0.001 versus GFAP-LepR+/+. Results are means ± the s.e.m. P values for unpaired comparisons were analyzed by two-tailed Student's t-test.
Supplementary Figure 7 Metabolic phenotypes of GFAP-LepR+/+ and GFAP-LepR–/–mice.
Five-week-old male mice were administrated tamoxifen (Tx) or vehicle (Veh) and then analyzed metabolic phenotype at three-month-old age. GFAP-LepR–/–mice did not show significant changes of (a) body weight (n=6 mice for GFAP-LepR+/+-veh; n=8 mice for GFAP-LepR+/+-tx; n=9 mice for GFAP-LepR–/–-tx, p=0.3720, t(12)=0.9274 for GFAP-LepR+/+-veh versus GFAP-LepR+/+-tx; p=0.08, t(15)=1.874 for GFAP-LepR+/+-tx versus GFAP-LepR–/–-tx), (b) fat mass (n=6 mice for GFAP-LepR+/+-veh; n=6 mice for GFAP-LepR+/+-tx; n=5 mice for GFAP-LepR–/–-tx, p=0.588, t(10)=0.5630 for GFAP-LepR+/+-veh versus GFAP-LepR+/+-tx; p=0.1974, t(9)=1.392 for GFAP-LepR+/+-tx versus GFAP-LepR–/–-tx) (c) lean mass (n=6 mice for GFAP-LepR+/+-veh; n=6 mice for GFAP-LepR+/+-tx; n=5 mice for GFAP-LepR–/–-tx, p=0.405, t(10)=0.8686 for GFAP-LepR+/+-veh versus GFAP-LepR+/+-tx; p=0.1410, t(9)=1.614 for GFAP-LepR+/+-tx versus GFAP-LepR–/–-tx) (d) food intake (n=5 mice for GFAP-LepR+/+-veh; n=7 mice for GFAP-LepR+/+-tx; n=7 mice for GFAP-LepR–/–-tx, p=0.8471, t(10)=0.1979 for GFAP-LepR+/+-veh versus GFAP-LepR+/+-tx; p=0.6162, t(12)=0.05146 for GFAP-LepR+/+-tx versus GFAP-LepR–/–-tx) (e, f) energy expenditure (n=8 mice for GFAP-LepR+/+; n=8 mice for GFAP-LepR–/–, p=0.8378, t(14)=0.2086 for light period; p=0.8641, t(14)=0.1743 for dark period; p=0.9966, t(14)=0.004295 for total) and (g, h) physical activity (n=8 mice for GFAP-LepR+/+; n=8 mice for GFAP-LepR–/–, p=0.1184, t(14)=1.663 for light period; p=0.2703, t(14)=1.148 for dark period; p=0.1692, t(14)=1.149 for total) when compared to littermate control mice. Results are means ± the s.e.m. P values for unpaired comparisons were analyzed by two-tailed Student's t-test.
Supplementary Figure 8 Representative images of Fos-positive POMC or AgRP neurons in the Arc of GFAP-LepR+/+ and GFAP-LepR–/– mice.
(a) Representative images show double labeled POMC-GFP and Fos cells in the Arc of GFAP-LepR+/+ and GFAP-LepR–/– mice.Scale bar = 100 μm. (b) Representative images show double labeled AgRP-GFP and Fos cells in the Arc of GFAP-LepR+/+ and GFAP-LepR–/– mice. Scale bar = 100 μm.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–8 (PDF 1619 kb)
Rights and permissions
About this article

Cite this article
Kim, J., Suyama, S., Koch, M. et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci 17, 908–910 (2014). https://doi.org/10.1038/nn.3725
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.3725
This article is cited by
-
Hypothalamic astrocyte NAD+ salvage pathway mediates the coupling of dietary fat overconsumption in a mouse model of obesity
Nature Communications (2024)
-
Hypothalamic GABRA5-positive neurons control obesity via astrocytic GABA
Nature Metabolism (2023)
-
Deletion of endothelial leptin receptors in mice promotes diet-induced obesity
Scientific Reports (2023)
-
A conceptual framework for astrocyte function
Nature Neuroscience (2023)
-
Proliferative Markers and Neural Stem Cells Markers in Tanycytes of the Third Cerebral Ventricle in Rats
Bulletin of Experimental Biology and Medicine (2023)
