
Abstract
Approximately 20 % of individuals with Parkinson's disease (PD) report a positive family history. Yet, a large portion of causal and disease-modifying variants is still unknown. We used exome sequencing in two affected individuals from a family with late-onset PD to identify 15 potentially causal variants. Segregation analysis and frequency assessment in 862 PD cases and 1,014 ethnically matched controls highlighted variants in EEF1D and LRRK1 as the best candidates. Mutation screening of the coding regions of these genes in 862 cases and 1,014 controls revealed several novel non-synonymous variants in both genes in cases and controls. An in silico multi-model bioinformatics analysis was used to prioritize identified variants in LRRK1 for functional follow-up. However, protein expression, subcellular localization, and cell viability were not affected by the identified variants. Although it has yet to be proven conclusively that variants in LRRK1 are indeed causative of PD, our data strengthen a possible role for LRRK1 in addition to LRRK2 in the genetic underpinnings of PD but, at the same time, highlight the difficulties encountered in the study of rare variants identified by next-generation sequencing in diseases with autosomal dominant or complex patterns of inheritance.
Similar content being viewed by others

Introduction
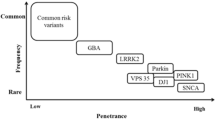
Characterized by resting tremor, bradykinesia, rigidity, and postural instability, Parkinson's disease (PD) is a prominent neurodegenerative disorder. Genetic factors contribute to the risk of PD—both sporadic and familial. Although up to 20 % of PD cases are believed to be familial [1, 2], thus far, rare genetic variants in only a few genes have been unequivocally shown to underlie these familial forms. They include PARK2/PARKIN, PINK1, PARK7/DJ-1, SNCA, and LRRK2 [3–8]. While all of these were identified by classical linkage analysis in large, multi-generation families, recently, next-generation sequencing has enabled the identification of disease-causing variants in smaller families and—what is especially important with regard to the investigation of neurodegenerative conditions with an onset late in life—without the need of genotypic information from more than one generation of affected individuals. Recently, exome sequencing was used to identify VPS35 as an additional gene involved in late-onset familial PD [9, 10]. Still, to date, the identified genes only explain a small portion of the genetic burden in familial PD. It is likely that genetic factors involved in bringing about the PD phenotype comprise both genetic variants of strong effect as well as variants of weaker effect which contribute to disease risk or phenotypic modification. A thorough understanding of the entire spectrum of genetic alterations implicated in the disease is necessary to better understand disease pathogenesis and to provide more specific treatment options in the future.
Here, we describe whole exome sequencing in a German family with autosomal dominant late-onset PD in whom known PD-linked mutations has previously been excluded in an attempt to pinpoint the disease-causing genetic variant. Two variants in leucine-rich repeat kinase 1 (LRRK1) and eukaryotic translation elongation factor 1 delta (EEF1D) emerged as the best candidate variants.
Materials and methods
Participants
The family was evaluated by neurologists specializing in movement disorders. All family members received a detailed neurologic exam. Information on deceased family members was gathered from medical and family records. Cases and controls used in genotyping and variant screening have been reported previously [9, 11] and are described in more detail in the supplement. Ethics review board approval and participants' written informed consent were obtained prior to the initiation of the study.
Analysis of copy number variation
Genome-wide copy number variant (CNV) analysis was carried out using Affymetrix Whole-Genome 2.7 M Array in conjunction with the Chromosome Analysis Suite with a confidence index of 85, a minimum homozygous region size of 10 kb and a minimum probe count of 5.
Exome sequencing
Exome sequencing was performed on a Genome Analyzer IIx (Illumina) after in-solution enrichment of exonic sequences (SureSelect Human All Exon 38 Mb kit, Agilent). For both samples, two lanes of a flow cell were sequenced, each as 54-bp paired-end runs. Read alignment was carried out with BWA (version 0.5.8) to the human genome assembly hg19. Single nucleotide variants (SNVs) and small insertions and deletions (indels) were detected with SAMtools (version 0.1.7). Prior to exome sequencing, presumably causal mutations in known Parkinson's disease genes (SNCA, PARK2, DJ1, PINK1, and LRRK2 (p.G2019S only)) had been excluded. Moreover, no known PD-linked variants were identified in either V:8 or V:17 by exome sequencing.
Genotyping
All 15 candidate variants were genotyped in 862 cases (376 of German (age 71.1 ± 9.4 years, 31.6 % female) and 486 of Austrian (age 58.7 ± 11.3 years, 35.4 % female) origin) and 1,014 population-based controls pertaining to the KORA-AGE cohort (age 76 ± 6.6 years, 50.1 % female) using MALDI-TOF mass spectrometry on the Sequenom® platform. Association was tested using the allelic test in PLINK.
Variant screening
We used Idaho®'s LightScanner high-resolution melting curve analysis to screen the eight coding exons of EEF1D for variants in the same set of 862 cases and 1,014 controls. For technical reasons, a part of exon 3 of EEF1D could not be evaluated. For LRRK1, the ras of complex proteins (ROC, p.631 to 826), the C-terminal of ROC (COR, p.827 to 1241), and the kinase (p.1242 to 1525) domains as determined by an InterproScan sequence search or extracted from the literature [12] were screened. In the case of altered melting patterns suggestive of variants, Sanger sequencing ensued. Significance was judged using the χ2 test.
Bioinformatic prioritization of variants
We collected a set of reference SNVs known to impair LRRK1 function. After computing a multiple sequence alignment using ClustalW based on LRRK1/LRRK2 pairs in 18 organisms, we introduced mutations into LRRK1 which mimic non-synonymous LRRK2 mutations related to PD (rs33939927 (p.Arg1441Gly), rs35801418 (p.Tyr1699Cys), rs34637584 (p.Gly2019Ser), rs35870237 (p.Ile2020Thr)), and added LRRK1 variants with a reported functional impact [12] to the set of reference SNVs. An in silico approach was applied to determine the disease potential of reference SNVs and the novel, non-synonymous LRRK1 variants. To reduce the error rates of single models in predicting the functional effect of a given variant on the protein, we implemented a multi-model ensemble combining prediction results of six publically available prediction algorithms into a combined Pscore (Fig. 3a, online methods). Additionally, a Dscore was computed, scoring the severity of structural changes between the wild type and the variant peptide based on the mean square deviation (online methods). By combining the Pscore and the Dscore, we computed a single overall mutation score (Mscore), rating the disease potential of an SNV between 0 (harmless polymorphism) and 1 (disease mutation) (online methods). SNVs were then ranked by their Mscore, and hierarchical clustering was conducted by Ward's minimum variance agglomeration method and Euclidean distance matrix and analyzed in R; p values were calculated by multi-scale bootstrap resampling [13]. Also see supplement.
Cellular analyses
Cellular analyses were carried out as previously described [14]. For a detailed description, see supplement.
Results
Pedigree and clinical phenotype
We describe a five-generation family from Southern Germany in which six members were affected by PD and the pattern of inheritance seems to be autosomal dominant with reduced penetrance (Fig. 1). Clinical assessment revealed a tremor-dominant, levodopa-responsive Parkinson's syndrome with an age of onset at 56.7 ± 1.15 years in all living affected individuals (V:8, V:9, and V:17, Online Resources Tabl 1). Further, all three affected individuals also showed positive Babinski signs and suffered episodes of depression. Mild to moderate cognitive impairment especially with regard to visuoconstruction, memory, and attention was present in all individuals. Dopamine transporter SPECT (DAT-SPECT) performed in two affected individuals (V:8 and V:17) revealed reduced tracer uptake in the putamen and asymmetrically in the caudate nucleus, in line with a diagnosis of PD.

Pedigree of family used for exome sequencing. Open symbols indicate unaffected family members; affected individuals are denoted by closed symbols. An arrow denotes the proband. Sex was obscured and birth order was altered to protect privacy. A diagonal line indicates a deceased individual
The affected parent and aunt (IV:5 and IV:7) of the proband died before initiation of the study. An additional cousin, V:1, had Parkinson's syndrome but also suffered from multiple sclerosis. She also died before initiation of the study. Lastly, a second cousin removed by four generations is also known to suffer from late-onset PD. The prevalence of PD in the general population is approximately 1 % [15]. Accordingly, we expected to find at least one phenocopy in this extended pedigree of 114 individuals. Since no additional family members on her side of the family showed signs of PD and since she shared none of the candidate variants common to the other three affected individuals examined, we concluded that it is unlikely that PD in her case is due to the same genetic variant as in the other affected individuals.
Identification of candidate variants by exome sequencing and segregation analysis
A genome-wide CNV scan revealed no structural variation ≥ 10 kb common to two affected members of the family (V:8 and V:17, Fig. 1). Exome sequencing was performed for the same individuals. This generated 6.57 gigabases (Gb) of alignable sequence for V:8 (average coverage = 70.93, >8× coverage = 90.65 %) and 6.67 Gb for V:17 (average coverage = 76.29, >8× coverage = 92.23 %). All detected variants shared by the two affected individuals (16,283 variants) were filtered against variants annotated in dbSNP132 as well as in-house exomes (n = 1076) of individuals with unrelated diseases and variants with a minor allele frequency (MAF) ≥ 0.01 were excluded from the follow-up, leaving 71 coding variants. Of these, 36 variants were predicted to alter the amino acid sequence (i.e., missense, nonsense, stop-loss, splice site, or frameshift variants and indels) and were genotyped in a third affected individual (V:9) (Online Resources Fig 1). Fifteen variants in 15 genes were present in all three affected individuals and were pursued further by Sanger sequencing-based testing for segregation in 32 members of the family belonging to generation V. Under the assumption that a given variant would be causal for PD, penetrances ranged between 30 and 50 %, with variants in LRRK1, EEF1D, and ARHGAP39 reaching the highest predicted penetrances (Table 1).
Frequency assessment of candidate variants in a case/control cohort
We genotyped the remaining 15 variants in a case/control sample, consisting of 862 individuals with PD and 1,014 KORA-AGE general population controls (Table 1). Two assays (UGT1A9 p.Val167Ala, TUBB6 p.Thr275Ala) did not meet quality control thresholds and were excluded from the analysis. The remaining 13 variants were, overall, very rare. Six (EEF1D p.Ala549Val, MUC17 p.Gln4310X, CCDC60 p.Arg155His, NAAA p.Arg211Trp, PTPRN2 p.Glu317Lys, and GLP2R p.Ile61Met) were validated in the proband but were otherwise not found again in the 1,876 individuals tested. FCGBP p.Glu4657fs was present in one additional PD patient but not in controls, and ZNF438 p.Thr454Ile was found in the proband and one control. Four additional variants annotated in dbSNP132 were identified at similar frequencies in cases and controls (ARHGAP39 p.Arg667Gln and MFSD3 p.Met311Thr) or were more common in controls than in cases (AQP4 p.Met202Thr and BRCA2 p.The1524Val). LRRK1 p.Arg1261Gln was found in eight controls and in four cases of our case/control sample (MAF 0.23/0.40 %). However, in four other control samples (680 additional KORA general population controls (0.07 %), 1,076 in-house exomes (0.05 %), 1,000 genomes (0.00 %), and NHLBI-ESP exomes (0.09 %)), MAFs were significantly lower, and the variant was, therefore, also analyzed further.
Mutational screening of EEF1D and LRRK1 in case/control cohort
While no single clear candidate for a causal variant emerged, two genes—EEF1D and LRRK1—were interesting with regard to functional considerations and predicted penetrance for PD in the family. The translation machinery has recently been implicated in PD pathogenesis [16, 17]. Also, the EEF1D p.Ala549Val variant was not found again in 3,064 individuals (genotyping cohort plus in-house exomes) and was also not annotated in the 1000 Genomes database. LRRK1, the paralog of the well-established PD gene LRRK2, has been shown to regulate endosomal protein transport, thus linking it to the lysosomal pathway [18] which may be compromised in PD [19, 20]. Formation of heterodimers between LRRK1 and LRRK2 has also been reported [21, 22]. We screened the coding regions of these genes in 862 Austrian and German PD cases and 1,014 controls searching for additional variants. This cohort comprised the same individuals used for the above frequency assessment of exome variants. We identified seven (six non-synonymous, one del) novel variants predicted to change the amino acid sequence of EEF1D. These were rare and occurred with similar frequencies in cases (five individuals with a variant) and controls (four individuals with a variant) (p > 0.5, χ2 test, Online Resources Tab 2). Variants did not cluster in a specific part of the gene (Fig. 2). The ROC, COR, and kinase domains of LRRK1 harbored a total of 20 novel amino acid sequence-changing variants (19 non-synonymous, 1 del) and 2 previously reported non-synonymous variants (rs56003881, rs41531245). Variants were found at similar frequencies in both groups (30 in cases, 31 in controls) (p > 0.5, χ2 test, Online Resources Tab 2). While small numbers preclude quantitative analyses, it is noteworthy that within the first 20 bp of the kinase domain, variants were present in both cases and controls, while beyond p.1262, all non-synonymous variants identified in the kinase domain occurred in cases only (Fig. 2). None of the individuals harboring LRRK1 variants were also positive for known LRRK2 variants p.Arg1441Cys, p.Tyr1699Cys, p.Gly2019Ser, or p.Ile2020Thr.

Location of EEF1D and LRRK1 variants identified in variant screening in relation to known functional domains. An asterisk denotes the variant identified by exome sequencing Variants printed in blue and annotated above the gene were found in cases, variants in green and below the gene were found in controls
Prioritization of LRRK1 variants using a novel bioinformatics algorithm
Since heterodimer formation between LRRK1 and LRRK2 has been described [21, 22], we decided to further assess the identified variants in LRRK1. To this end, we used a novel bioinformatics algorithm based on a multi-model ensemble of prediction algorithms and structural analysis to select variants in LRRK1 for functional follow-up. Mutation scores were calculated for the 19 novel, non-synonymous LRRK1 variants identified in both cases and controls, the LRRK1 variants (p.Lys746Glu, p.Phe1022Cys, p.Gly1411Arg and p.Ile1412Thr) corresponding to four known pathogenic LRRK2 mutations (p.Arg1441Gly, p.Tyr1699Cys, p.Gly2019Ser, and p.Ile2020Thr) and three artificial variants known to abolish LRRK1 GTP-binding (p.Lys651Ala) and kinase activity (p.Lys746Gly and p.Lys1270Trp) (Fig. 3a) [12]. Hierarchical clustering showed that three of the novel variants (p.Arg631Trp, p.Arg1271His and p.Tyr1410Asp)—present only in PD cases—clustered with the LRRK1 equivalents of LRRK2 p.Arg1441Gly, p.Tyr1699Cys, p.Gly2019Ser, and p.Ile2020Thr as well as the kinase- and GTP-binding dead amino acid substitutions (Fig. 3b). Accordingly, these three variants in addition to the initial variant identified by exome sequencing (p.Arg1261Gln) were selected for functional follow-up.

Prediction of pathogenic potential of newly identified variants. a For each variant (colored lines) the predicted score s of an individual algorithm, its reliability r, and the transformed score p(s, r, c) are shown. Variants holding a predicted disease-causing potential (class = 1) were respectively marked with an asterisk. The diverse results among each single algorithm motivated the calculation of one combined score (Pscore), which was adjusted by additional structural analyses (Dscore) resulting in a mutation score (Mscore). The highest scoring variant is p.Tyr1410Asp (Mscore = 0.839), a variant only present in PD cases, followed by the LRRK2 equivalent of Gly2019Ser (Mscore = 0.771), the loss of autophosphorylation mutation Lys1270Trp (Mscore = 0.768), and two variants abolishing kinase activity: Ile1412Thr (Mscore = 0.728) and Lys746Gly (Mscore = 0.723). b Hierarchical clustering with Ward's minimum variance agglomeration method and Euclidean distance matrix shows that three of the novel variants which were only found in individuals with PD (p.Arg631Trp, p.Arg1271His, and p.Tyr1410Asp) cluster with the LRRK1 equivalents of LRRK2 p.Arg1441Gly, p.Tyr1699Cys, p.Gly2019Ser, and p.Ile2020Thr as well as the LRRK1 kinase- and GTP-binding dead amino acid substitutions
Functional assessment of LRRK1 variants
In SHSY5Y neuroblastoma cells, levels of protein expression as assessed by Western blot were not changed by any of the four newly identified variants or the artificial variants ablating GTP-binding (p.Lys651Ala) or kinase activity (p.Lys1270Trp) (Fig. 4a). Likewise, the presence of these variants was not associated with significant toxicity as measured by MTT assay (Fig. 4b) and did not alter cytoplasmic localization of myc-tagged LRRK1 (Fig. 4c). Like others [23, 24], we could not detect LRRK1 kinase activity above the background and could not determine whether activity was altered by the variants.

Cellular expression of LRRK1 and mutant variants. a Western blot analysis of myc-tagged LRRK1 expression in SHSY5Y cells with beta actin-loading control. b Analysis of LRRK1 toxicity as measured by MTT assay in SHSY5Y cells. No significant toxicity was associated with wild-type LRRK1, artificial mutations in LRRK1, or disease-associated coding changes. Data is expressed as percentage of untransfected control cells, mean, and standard error measurement displayed. c Immunocytochemistry analysis of myc-tagged LRRK1 constructs. Staining for myc is shown separately and merged. All tagged constructs displayed a diffuse cytoplasmic staining pattern. Scale bar = 20 μm
Discussion
In an unbiased, whole exome approach, we identified a variant in LRRK1 (p.Arg1261Gln) as a candidate for a potentially causal variant in familial PD. Although this finding is intriguing and functionally plausible, we are unable to conclude that this is indeed the cause of PD in our family. For one, the variant was found in both cases and controls in our larger case/control sample. Yet, the actual variant frequency in controls appears to be lower than that found in the KORA-AGE cohort (8 in 1,014 KORA-AGE vs. 0 in 1,000 genomes, 1 in 1,076 in-house exomes, and 1 in additional 680 KORA controls), and it could be possible that KORA-AGE is enriched for the LRRK1 p.Arg1261Gln variant due to a founder effect or that is also present in controls because its PD-related nature depends on a specific genetic context. Secondly, the other 14 identified rare variants shared by all three affected individuals also represent potential candidates. Especially, EEF1D p.Ala549Val, which was not found again in any individual genotyped (n = 3064, case/control sample and in-house exomes) or the 1,000 genomes or the NHLBI-ESP exomes, represents another good contender. In general, these findings draw attention to the fact that in many cases, very large populations will need to be evaluated to conclusively judge the disease-related nature of a rare variant such as those identified by exome sequencing. Most recent studies show that while the power to detect associations for genes harboring rare variants varies widely across genes, only <5 % of genes achieved 80 % power even assuming high odds ratios (OR) of 5 when tested in 400 cases and 400 controls [25]. Ultimately, it is also possible that the truly causal variant was not picked up in this study because it lies outside the targeted regions of the exome.
The fact that both the LRRK1 and EEF1D variants were also found in three unaffected members of the family each per se does not contradict potential causality as it is known from other autosomal dominant forms of PD that even among members of a single family, penetrance of known PD mutations can vary widely. Of individuals who harbor the LRRK2 p.Gly2019Ser mutation, for example, only 28 % will develop PD by the age of 59 years [26]. Thus, predicted penetrances of the variants identified in our family are in line with what is reported in the literature for other forms of autosomal dominant PD.
Both LRRK1 and LRRK2 belong to the Roco family of proteins. These proteins are likely to perform a number of different functions as they are not only characterized by a conserved Ras-like GTPase domain called ROC and a characteristic COR domain of unknown function but also harbor kinase and protein–protein interaction domains [27]. While a contribution of mutations in LRRK2 to disease development in PD seems firmly established, the role of LRRK2 paralog LRRK1 is unclear. It is known that LRRK1 and LRRK2 form heterodimers in HEK293T cells [21, 22] and that both proteins are expressed in similar tissues. Accordingly, a hypothetical role for LRRK1 in addition to LRRK2 is plausible.
The precise role of LRRK2 in PD pathogenesis, however, has not been fully established. Accordingly, even if one were to assume a similar role of LRRK1 in disease development, exactly which function of the protein would be involved in the disease is uncertain. Therefore, the lack of a functional effect on protein expression levels, subcellular localization, and cell viability of the LRRK1 variants we identified does not equate to a definitely missing role of LRRK1 in PD. Interestingly, Lrrk1 has also been implicated in a quantitative trait locus for dopaminergic amacrine cell number in the murine retina [28]. Further, the recent link between LRRK1 and endosomal protein trafficking [18, 29] is also very intriguing in light of the fact that one of the postulated pathomechanisms for LRRK2 in PD involves aberrant lysosomal function or localization [20, 30, 31].
However, studies addressing the role of both common and rare genetic variants in LRRK1 with regard to PD do not seem to substantiate the conception of LRRK1 as a “PD gene” [21, 32–34]. While none of these studies found a common or rare variant clearly linked to PD, nonetheless, across three studies ([32, 34] and our study), the p.Thr967Met variant has only been identified in seven out of 1,552 cases but not in any of 1,535 controls (p nominal ≤ 0.01, χ2 test; not significant after correction for multiple testing, OR = 14.90 (95 % confidence interval = 0.85 to 261.18)). Yet, in a family with multiple individuals with PD, the variant did not segregate with the phenotype [34]. Evidence also suggests that variants in LRRK1 are able to modify the PD phenotype. Tunisian individuals with LRRK2 p.Gly2019Ser showed a trend towards a 6-year earlier age of onset when they also carried LRRK1 p.Leu416Met [20]. In line with this, it has been demonstrated for other genetic disorders that genetic variants at related loci can both drive and modify a given phenotype depending on the variant and the genetic context [35]. At the moment, both functional and genetic data addressing a role of LRRK1 as a PD gene are inconclusive. Nonetheless, it is interesting that in our unbiased whole exome approach, one of the top candidate variants for a genetic factor underlying or contributing to the PD phenotype in our family is a non-synonymous variant in the kinase domain of LRRK1 and that other individuals suffering from PD harbor LRRK1 variants (p.Tyr1410Asp) only with one amino acid away from the location which is equivalent to the prominent p.Gly2019Ser mutation of LRRK2.
In summary, all variants shared by the three affected individuals in our family represent potential causal or modifying alleles in PD. As is the case for all rare and very rare variants, establishing definitive causality is difficult, and only the identification of additional PD families harboring these variants or their analysis in sufficiently powered case/control studies will tell whether these variants do indeed hold a role in bringing about PD.
References
Payami H, Larsen K, Bernard S, Nutt J (1994) Increased risk of Parkinson's disease in parents and siblings of patients. Ann Neurol 36:659–661
Bonifati V, Fabrizio E, Vanacore N, De Mari M, Meco G (1995) Familial Parkinson's disease: a clinical genetic analysis. Can J Neurol Sci 22:272–279
Zimprich A, Biskup S, Leitner P et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607
Bonifati V, Rizzu P, van Baren MJ et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259
Kitada T, Asakawa S, Hattori N et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Paisan-Ruiz C, Jain S, Evans EW et al (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44:595–600
Polymeropoulos MH, Lavedan C, Leroy E et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047
Valente EM, Abou-Sleiman PM, Caputo V et al (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304:1158–1160
Zimprich A, Benet-Pages A, Struhal W et al (2011) A Mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89:168–175
Vilarino-Güell C, Wider C, Ross OA et al (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89:162–167
Wichmann HE, Gieger C, Illig T (2004) KORAgen—resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen 67(Suppl 1):26–30
Korr D, Toschi L, Donner P, Pohlenz HD, Kreft B, Weiss B (2006) LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal 18:910–920
Shimodaira H (2004) Approximately unbiased tests of regions using multistep-multiscale bootstrap resampling. Ann Statistics 32:2616–2641
Greggio E, Lewis PA, van der Brug MP et al (2007) Mutations in LRRK2/dardarin associated with Parkinson disease are more toxic than equivalent mutations in the homologous kinase LRRK1. J Neurochem 102:93–102
Elbaz A, Bower JH, Maraganore DM et al (2002) Risk tables for parkinsonism and Parkinson's disease. J Clin Epidemiol 55:25–31
Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C et al (2011) Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet 89:398–406
Gehrke S, Imai Y, Sokol N, Lu B (2010) Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 466:637–641
Hanafusa H, Ishikawa K, Kedashiro S et al (2011) Leucine-rich repeat kinase LRRK1 regulates endosomal trafficking of the EGF receptor. Nat Comm 2:158
Sidransky E, Nalls MA, Aasly JO et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 361:1651–1661
Tong Y, Yamaguchi H, Giaime E et al (2010) Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A 107:9879–9884
Dachsel JC, Nishioka K, Vilarino-Güell C et al (2010) Heterodimerization of Lrrk1-Lrrk2: implications for LRRK2-associated Parkinson disease. Mech Age Dev 131:210–214
Klein CL, Rovelli G, Springer W, Schall C, Gasser T, Kahle PJ (2009) Homo- and heterodimerization of ROCO kinases: LRRK2 kinase inhibition by the LRRK2 ROCO fragment. J Neurochem 111:703–715
Deng X, Dzamko N, Prescott A et al (2011) Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol 7:203–205
Civiero L, Vancraenenbroeck R, Belluzzi E et al (2012) Biochemical characterization of highly purified leucine-rich repeat kinases 1 and 2 demonstrates formation of homodimers. PLoS One 7:e43472
Tennessen JA, Bigham AW, O’Connor T et al (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337:64–69
Healy DG, Falchi M, O’Sullivan SS et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case–control study. Lancet Neurol 7:583–590
Marin I, van Egmond WN, van Haastert PJM (2008) The Roco protein family: a functional perspective. FASEB 22:3103–3110
Whitney IE, Raven MA, Ciobanu DC, Williams RW, Reese BE (2009) Multiple genes on chromosome 7 regulate dopaminergic amacrine cell number in the mouse retina. Invest Ophthal Vis Sci 50:1996–2003
Ishikawa K, Nara A, Matsumoto K, Hanafusa H (2012) EGFR-dependent phosphorylation of leucine-rich repeat kinase LRRK1 is important for proper endosomal trafficking of EGFR. Mol Biol Cell 23:1294–1306
Gómez-Suaga P, Luzón-Toro B, Churamani D et al (2012) Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet 21:511–525
Dodson MW, Zhang T, Jiang C, Chen S, Guo M (2012) Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet 21:1350–1363
Haugarvoll K, Toft M, Ross OA, White LR, Aasley JO, Farrer MJ (2007) Variants in the LRRK1 gene and susceptibility to Parkinson's disease in Norway. Neurosci Lett 416:299–301
Chung SJ, Armasu SM, Biernacka JM et al (2011) Common variants in PARK loci and related genes and Parkinson's disease. Mov Disord 26:280–288
Taylor JP, Hulihan MM, Kachergus JM et al (2007) Leucine-rich repeat kinase 1: a paralog of LRRK2 and a candidate gene for Parkinson's disease. Neurogenetics 8:95–102
Davis EE, Zhang Q, Liu Q et al (2011) TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet 43:189–196
Acknowledgments
We are gratefully indebted to Katja Junghans, Susanne Lindhof, Jelena Golic, Sybille Frischholz, and Regina Feldmann at the Institut für Humangenetik, Helmholtz Zentrum München, Munich, Germany, for their expert technical assistance in performing Sequenom genotyping and Light Scanner analyses. This study was funded by in-house institutional funding from Technische Universität München and Helmholtz Zentrum München, Munich, Germany. Recruitment of case and control cohorts was supported by institutional (HelmholtzZentrumMünchen, Munich, Germany) and government funding from the German Bundesministerium für Bildung und Forschung (03.2007-02.2011 FKZ 01ET0713). PAL is funded by a Parkinson's UK research fellowship (F1002). The work performed by PAL, SD, and CM was also funded in part by grants from the Michael J. Fox Foundation LRRK2 consortium and by the Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698) to the UK Parkinson's Disease Consortium (UKPDC) whose members are from the UCL Institute of Neurology, the University of Sheffield, and the MRC Protein Phosphorylation Unit at the University of Dundee.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 209 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Schulte, E.C., Ellwanger, D.C., Dihanich, S. et al. Rare variants in LRRK1 and Parkinson's disease. Neurogenetics 15, 49–57 (2014). https://doi.org/10.1007/s10048-013-0383-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-013-0383-8