



Abstract
Ataxia-telangiectasia (A-T) is a genetic disorder caused by a mutation of the Atm gene, which controls DNA repair, cell cycling, and redox homeostasis. Even though oxidative stress has been implicated in the neurological anomalies in A-T, the effects of ATM loss on neural stem cell (NSC) survival has remained elusive. In this study, we investigated the effects of oxidative stress on NSC proliferation in an animal model for A-T neurodegeneration. We found that cultured subventricular zone neurosphere cells from Atm−/− mice show impaired proliferation, as well as intrinsic elevation of reactive oxygen species (ROS) levels, compared with those from Atm+/+ mice. We also show that increasing the levels of ROS by H2O2 treatment significantly reduces Atm+/+ neurosphere formation and proliferation. In Atm−/− neurosphere cells, the Akt and Erk1/2 pathways are disrupted, together with enhanced activity of the p38 mitogen-activated protein kinase (MAPK). Treatment of these cells with the antioxidant N-acetyl-L-cysteine (NAC) or with a p38 MAPK inhibitor restores normal proliferation and reduced expression of p21cip1 and p27kip1 in the Atm−/− NSCs. These observations indicate that ATM plays a crucial role in NSC proliferation, by activating Akt and Erk1/2 pathways and by suppressing ROS-p38 MAPK signaling. Together, our results suggest that p38 MAPK signaling acts as a negative regulator of NSC proliferation in response to oxidative stress. These findings suggest a potential mechanism for neuronal cell loss as a result of oxidative stress in NSCs in progressive neurodegenerative diseases such as A-T.
Disclosure of potential conflicts of interest is found at the end of this article.
Introduction
In ataxia-telangiectasia (A-T), a disease characterized by progressive neurodegeneration, the Atm gene is mutated. Humans with A-T display progressive neuromotor deficits, with development of ataxia at an early age. In the brain, the cerebellum is initially the area most prominently affected, but eventually nearly all the regions of the brain show abnormalities [1, 2]. The ATM protein, encoded by the Atm gene, participates in regulating cell cycling in response to DNA damage and oxidative stress [3, 4]. ATM also modulates intracellular reactive oxygen species (ROS) levels, and when in excess, contributes to the neurodegeneration seen in A-T, caused by loss of ATM [5–7]. In light of this information, it is not surprising that treatment with antioxidants prevents Purkinje cell loss [8] and corrects neurobehavioral deficits of Atm−/− mice [9, 10]. However, it remains unclear how the loss of ATM results in the neurodegenerative phenotype seen in A-T.
In the normal mouse brain, it has been reported that ATM expression is abundant in NSC/neural progenitor cells (NPCs), but it is markedly downregulated as the cells differentiate. In the absence of ATM, abnormal differentiation occurs [11]. These findings suggest that ATM may play essential roles in NSC maintenance and differentiation. NSCs undergo asymmetric self-renewing division to produce neuronal and glial progenitor cells, which differentiate to become neurons, astrocytes, and oligodendrocytes. For this reason, proper control of NSC self-renewal and proliferation is crucial for the maintenance of neural homeostasis [12]. As noted above, despite the known role of ATM in redox homeostasis and potential role of ATM in NSC functions, the effects of ATM deficiency on NSC survival/proliferation and its mechanism remain poorly understood. It is important to note, however, that ROS has antiproliferative effects in NSCs [13]. Thus, oxidative stress may contribute to the disruption of proliferation of NSCs. The aim of this study is to show that loss of ATM leads to oxidative stress in NSCs and to address the effects of ATM loss-mediated oxidative stress on NSC proliferation.
Both Akt and Erk1/2 seem to play important roles in NSC survival, but their levels are downregulated when the NSCs differentiate. Disruption of these pathways using specific inhibitors results in loss of NSC viability, suggesting that they participate in NSC survival and maintenance [14–17]. More recent studies have shown that p38 mitogen-activated protein kinase (MAPK) is involved in the regulation of neural plasticity and stress responses and that it functions as a negative regulator of NSC proliferation during early brain development [18, 19]. Based on these findings, we hypothesized that oxidative stress may participate in the control of NSC survival by modulating these signaling pathways. To test this idea, we used NSCs prepared from the subventricular zone (SVZ) of Atm+/+ and Atm−/− mice and compared their ROS levels and the p38 MAPK pathway, as well as their proliferation rate.
We report here that SVZ cells from both Atm+/+ and Atm−/− mice retain stem cell properties in vitro, in terms of multipotency and self-renewal, and that they generate neurospheres when cultured in the presence of epidermal growth factor (EGF) and fibroblast growth factor (FGF). For Atm−/− neurospheres, however, the absence of ATM results in defective NSC proliferation, with a concomitant disruption of the Akt and Erk1/2 pathways. The p38 MAPK pathway is also disturbed, indicating that all three of signaling pathways are crucial for the maintenance of pluripotency in NSCs. Our results identify the activation of the p38 MAPK pathway as a mechanism by which elevated ROS levels limit Atm−/− NSC proliferation and survival.
Materials and Methods
Mice
The Atm−/− mice in this study were originally generated by Dr. C. Barlow. They were purchased from the Jackson Laboratory (Bar Harbor, ME, http://www.jax.org). We genotyped offspring of Atm+/− breeding pairs by real-time polymerase chain reaction-based assays of mouse tail DNA. Littermates were used as controls in all experiments. Animal care was in accordance with The University of Texas M.D. Anderson Cancer Center guidelines for animal experiments.
NSC Isolation and Neurosphere Culture
Neurospheres were obtained from P1 pup SVZ and were maintained in culture essentially as reported [20]. The walls of the lateral ventricles were removed and enzymatically dissociated in Hank's balanced saline solution (HBSS) buffer (5 mM KCl, 124 mM NaCl, 3.2 mM MgCl2, 100 μM CaCl2, 26 mM NaHCO3, and 10 mM glucose) containing 1 mg/ml trypsin at 37°C for 10 minutes. The tissue was centrifuged at 750 rpm for 5 minutes in soybean trypsin inhibitor. The dispersed cells were resuspended in HBSS buffer containing 0.7 mg/ml RNase free DNase and enzymatically disaggregated for 5 minutes at room temperature. The dissociated cells were centrifuged, resuspended in neural basal medium (NBM) containing 20 ng/ml EGF, 10 ng/ml FGF, 2 mM glutamine, antibiotics (100 units/ml penicillin and 100 g/ml streptomycin), and 0.125 μg/ml fungizone, and maintained in an atmosphere of 5% CO2 at 37°C. After 1–2 days of incubation, the cells formed neurospheres. Subcultures were prepared every 4–5 days by centrifugation of the neurospheres and dissociation of the cells in 1 ml of trypsin; single-cell suspensions were replated in new culture dishes in fresh medium to obtain new neurospheres. Experiments were performed with cultured cells between passages 2 and 5.
Chemical Reagents
The phosphatidyl inositol 3-kinase (PI3K) inhibitor LY294001 (10 μM), the MAP/Erk kinase (MEK) inhibitor PD98059 (50 μM), and the p38 MAPK inhibitor SB203580 (10 μM) were purchased from BD Biosciences (San Diego, http://www.bdbiosciences.com). The antioxidant NAC (1 mM) was kindly provided by M. Yan (M.D. Anderson Cancer Center) [21].
Analysis of Intracellular ROS
Intracellular ROS levels were monitored by using 2′-7′-dichlorofluorescein diacetate (H2DCFDA), which forms the fluorescent compound dichlorofluorescein on oxidation with ROS. After 30 minutes of incubation with 10 μM H2DCFDA, cells were rinsed twice with phosphate-buffered saline (PBS), and their fluorescence was monitored by scanning the whole well using a NucleoCounter (New Brunswick Scientific, Edison, NJ, http://www.nbsc.com), set at excitation and emission wavelengths of 485 and 535 nm. All experiments were performed in triplicate wells, and each experiment was performed at least three times. Results are expressed as the mean ± SD. Atm+/+ and Atm−/− neurospheres were always derived from litters produced by the same pregnant female.
Cell Proliferation Assays
After trypsinizing neurospheres, single neural stem cells were resuspended in NBM containing EGF and FGF. The cells were seeded into 96-well plates at a density of 102 cells per well, to allow cell communication by paracrine/autocrine factors for physiological proliferation. The drugs under study were added when the plates were seeded. After incubation for 48 hours, the numbers of newly formed neurospheres per well were counted, using phase contrast microscopy. To measure neurosphere sizes, images were obtained from three separate fields per well, and the volumes of neurospheres were estimated using a microimage analysis system from Olympus. To measure total cell proliferation in neurospheres, cells in each well were trypsinized, and total cell numbers per well were counted. Alternatively, single cells in suspension were seeded into 96-well plates at a concentration of 102 cells per well. Each well was fed with [3H]-thymidine (2 μCi)/EGF/FGF–containing medium for 4 hours, before measurement of [3H]-thymidine incorporation into DNA.
Differentiation of Neural Stem Cells
Neurospheres were enzymatically dissociated as described above. The cells were seeded on to chamber slides and maintained in medium containing 10% fetal bovine serum (FBS), without EGF and FGF for 7 days. Antibodies used for characterization of neural stem cells were anti-Ki67 (Abcam Biotechnology, Cambridge, U.K., http://www.abcam.com); anti-Map2 (Cell Signaling Technology, Beverly, MA, http://www.cellsignal.com); anti-GFAP; and anti-nestin (Santa Cruz Biotechnology, Santa Cruz, CA, http://www.scbt.com).
Immunocytochemical Image Analysis
Neurospheres were seeded on 2- or 8-well chamber slides coated with poly-L-lysine/lamin and incubated for 3 hours to facilitate adhesion. These cells, which will be referred to as “adhered neurospheres” in the text, maintained the properties of undifferentiated cells, as evidenced by anti-nestin staining. Adhered neurospheres also respond to H2O2 in a manner similar to the floating neurospheres. For ROS response experiments, neurospheres in monolayer cultures were treated with H2O2 in NBM for 16 hours, after which the cells were fixed with 4% paraformaldehyde in PBS for 30 minutes at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 15 minutes. For staining with antibodies, the cells were preincubated for 2 hours at 37°C with 10% FBS in PBS and further incubated at 4°C overnight with primary antibodies. The antibody-bound cells were washed three times in PBS for 5 minutes each, and fluorescent secondary antibodies were added for 1 hour at 37°C. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The stained slides were mounted in Slowfade (Molecular Probes, Eugene, OR, http://www.probes.invitrogen.com). Cells were imaged under an Olympus IX2-SL microscope, equipped with a ×400 objective and an Olympus DP 70 digital camera connected to a PC computer (Olympus, Tokyo, http://www.olympus-global.com.
Protein Analysis
For protein measurement, cells were seeded into 10-cm dishes and treated with H2O2 at different concentrations. At indicated times after H2O2 treatment, cells were washed twice with ice-cold PBS and scraped directly into lysis buffer containing 150 mM NaCl, 0.5% wt/vol SDS, 0.5% vol/vol NP40, 0.5% wt/vol sodium deoxycholate, 1 mM EGTA, and a mixture of protease inhibitors (Complete Mini tablets; Boehringer Mannheim, Mannheim, Germany, http://www.boehringer.com). For Western blotting, protein concentrations were determined using a Bradford reagent (Bio-Rad, Hercules, CA, http://www.bio-rad.com). Proteins (30 μg) were separated by SDS-polyacrylamide gel electrophoresis on 10%-12% gels and transferred to polyvinylidene difluoride membranes before incubation with antibodies.
Antibodies
Antibodies used for Western blotting analysis were anti-phospho-Akt (Ser473), anti-Akt, anti-phospho-Erk1/2 (Thr202/Tyr204), anti-Erk1/2, anti-phospho-p38 (Th180/Tyr182), anti-p38, anti-p27kip1, anti-cyclin E, anti-cyclin D1, anti-superoxide dismutase 1 (SOD1), anti-caspase 3, anti-γ-H2AX (Cell Signaling Technology); anti-nestin, anti-p27kip1, anti-p21cip1, anti-p16INK4a, and anti-β-actin (Santa Cruz Biotechnology).
Apoptosis
Adhered cells grown for 16 hours on 8-well chamber slides coated with poly-L-lysine/lamin in NBM for 16 hours were fixed with 4% paraformaldehyde in PBS for 30 minutes at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 15 minutes. Detection of apoptotic cells was performed using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining, using a commercial kit (Roche Applied Science, Indianapolis, IN, http://www.roche-applied-science.com). Apoptotic cells were counted directly under a fluorescence microscope (IX2-SL; Olympus). Data are presented as the percentage of apoptotic cells in the total number of cells, which were counterstained with DAPI. A total of 10 fields were counted per slide.
Statistics
For each experiment, data are presented as the mean ± SE of values. Each experiment was repeated at least three times. Statistical comparisons of values for Atm+/+ versus Atm−/− neurospheres and for untreated control versus treated neurospheres were made using an analysis of variance, followed by Bonferroni's post hoc test. Differences were considered significant when p < .05. Analyses of data were performed using Prism 5 Software (GraphPad Software, San Diego, CA, http://www.graphpad.com).
Results
Characterization of Normal SVZ Neural Stem Cells In Vitro
NSCs are localized in the SVZ of the lateral ventricle and in the subgranular zone of the hippocampal dentate gyrus. Postnatal NSCs were isolated from the SVZ and cultured in the presence of EGF and FGF, as previously described [20]. These cells formed neurospheres in vitro cultures. After multiple subcultures, under these conditions, the neurosphere cultures maintain their stem cell characteristics. Fluorescence microscopy showed that the neurospheres were composed of nestin-positive cells and that most of them also expressed the cell cycling marker Ki67 (supporting information Fig. 1A). After 3 hours of attachment to slides coated with poly-L-lysine/lamin, the adhered cells still had stem cell-like phenotypes, expressing nestin and Ki67 (supporting information Fig. 1B). When the cells were cultured without EGF and FGF, however, their daughter cells differentiated into cells that expressed either glial (GFAP) or neuronal (Map2) markers, confirming that the neurosphere-repopulating cells are multipotent (supporting information Fig. 1C).
Decreased Proliferation in Atm−/− Neural Stem Cells Is Caused by Elevated ROS
To investigate the effects of ATM deficiency on NSC survival, we compared NSC proliferation in neurospheres prepared from Atm+/+ versus Atm−/− mice. After incubation of SVZ cells in the presence of EGF and FGF for 7 days, NSCs from both Atm+/+ and Atm−/− mice generated neurospheres. On visual inspection, no distinct differences were evident in primary cultured neurospheres from Atm+/+ versus Atm−/− mice. However, in secondary cultures, the Atm−/− neurospheres were fewer in number and were smaller than the Atm+/+ neurospheres. Figure 1A shows primary neurospheres from postnatal day 1 (p1) mouse brains of both genotypes (Atm+/+ and Atm−/−) and secondary neurospheres generated from primary neurospheres after subculturing. To compare the proliferative capacity of Atm+/+ versus Atm−/− NSCs, the numbers and sizes of new neurospheres generated 2 days after subculture and total numbers of cells per well were determined. The number of newly generated neurospheres is indicative of NSC self-renewing activity, the sizes of newly generated neurospheres represent proliferation within each neurosphere, and total cell numbers in a well represent mitotic division of daughter cells in neurospheres. As noted above, neurospheres from Atm−/− mice formed in significantly lower numbers and were significantly smaller than those from Atm+/+ mice. Furthermore, lower total cell numbers per well were present (Fig. 1B).
![Decreased proliferation in Atm−/− neural stem cells is caused by elevated ROS. (A): Phase-contrast photomicrographs of Atm+/+ and Atm−/− neurospheres. Primary neurospheres were obtained from p1 mouse brains, and secondary neurospheres were generated from primary neurospheres after subculture. Scale bars = 50 μm. (B): Numbers, sizes of newly formed neurospheres, and total cell numbers per well were assessed from in neural stem cells (NSCs) from Atm+/+ and Atm−/− mice. The mean ± SE of three independent experiments is shown. **, p < .01, when untreated Atm−/− NSCs were compared with untreated Atm+/+ NSCs. (C–G): NSCs were either left untreated or treated with NAC, and the following were assessed: (C) intracellular 2′-7′-dichlorofluorescein diacetate, (D) numbers of newly formed neurospheres, (E) size of newly formed neurospheres, (F) total cell numbers per well, and (G) [3H]-thymidine uptake. The mean ± SE of three independent experiments is shown. *, p < .05; **, p < .01, when untreated Atm−/− NSCs were compared with untreated Atm+/+ NSCs or when NAC-treated Atm−/− NSCs were compared with untreated Atm−/− NSCs. Abbreviations: NAC, N-acetyl-L-cysteine; ROS, reactive oxygen species.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/stmcls/27/8/10.1002_stem.125/3/m_stmcls_27_8_1987_nfig001.jpeg?Expires=1716312896&Signature=xIAYXu6VSK4Xa8XQQNHy8l7jgYQjeOwSclZC5rdw-CCz9jWGxWsvst-d0ULgXJq-Af~YoqPGGxlG5GR0vpovMRYA4pqUc4wt7rAgruWS-nxIa~Ijb4bVanuh9dl0RfzUbotSmTP5c9jlI0FyUWISdHE98vSvX6pEQpCF15hfvIvMYYwiHYV3LtMre49BC8I7ro7MzskgoKLhJNKE3b8DORDl3lUrzXpLL~ZLuw2Dx59fjYKpRqXxyM9vD2YHIAC31kiy6xYKOiAq9OrMiJ1MfYFS3KrGpKx0hkJ~scUWjLxjkI66ZKI5-VoSIhJWMOqo1vDUdji4xqpGP-uZtzwLZA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Decreased proliferation in Atm−/− neural stem cells is caused by elevated ROS. (A): Phase-contrast photomicrographs of Atm+/+ and Atm−/− neurospheres. Primary neurospheres were obtained from p1 mouse brains, and secondary neurospheres were generated from primary neurospheres after subculture. Scale bars = 50 μm. (B): Numbers, sizes of newly formed neurospheres, and total cell numbers per well were assessed from in neural stem cells (NSCs) from Atm+/+ and Atm−/− mice. The mean ± SE of three independent experiments is shown. **, p < .01, when untreated Atm−/− NSCs were compared with untreated Atm+/+ NSCs. (C–G): NSCs were either left untreated or treated with NAC, and the following were assessed: (C) intracellular 2′-7′-dichlorofluorescein diacetate, (D) numbers of newly formed neurospheres, (E) size of newly formed neurospheres, (F) total cell numbers per well, and (G) [3H]-thymidine uptake. The mean ± SE of three independent experiments is shown. *, p < .05; **, p < .01, when untreated Atm−/− NSCs were compared with untreated Atm+/+ NSCs or when NAC-treated Atm−/− NSCs were compared with untreated Atm−/− NSCs. Abbreviations: NAC, N-acetyl-L-cysteine; ROS, reactive oxygen species.
Brain cells of A-T children are under oxidative stress [4], and oxidative stress reduces proliferation of neural cells [22, 23]. To test whether loss of ATM leads to oxidative stress in NSCs, we measured intracellular ROS in NSCs derived from Atm+/+ versus Atm−/− SVZ, by measuring intracellular H2DCFDA fluorescence. Figure 1C shows that Atm−/− NSCs have higher ROS levels than Atm+/+ NSCs, but the antioxidant NAC significantly reduces ROS levels, making them similar to those of untreated Atm+/+ NSCs. To determine whether reduced proliferation in Atm−/− NSCs is caused by elevated ROS levels, Atm−/− NSCs were treated with NAC, and their proliferation rates were compared with those of untreated Atm−/− NSCs. Figure 1D–1F shows that NAC treatment of the cultures improved numbers, sizes, and total cell numbers per well of newly formed Atm−/− neurospheres. However, analysis of DNA synthesis by measuring incorporation of [3H]-thymidine into the cells indicated that NAC had only modest rescuing effects on DNA synthesis (Fig. 1G). This may be caused by contact inhibition occurring when NSCs form the neurosphere structure. Because the antioxidant NAC improved proliferation of Atm−/− NSCs to levels similar to those of Atm+/+ NSCs, these results indicate that NSCs require functional ATM, which modulates intracellular redox status for normal survival and proliferation.
H2O2-Treated Atm+/+ Neural Stem Cells Have Enhanced Intracellular ROS Levels and Show Reduced Proliferation
To substantiate our argument that elevated ROS levels cause defective NSC proliferation, Atm+/+ NSCs were treated with H2O2 at different concentrations, and their intracellular ROS levels were compared. H2DCFDA fluorescence intensity, indicative of ROS, increased in the presence of H2O2 in a concentration-dependent manner, but it was significantly reduced when the cultures were pretreated with NAC before exposure to H2O2 (Fig. 2A). We next investigated the effects of H2O2 treatment on DNA synthesis in Atm+/+ NSCs. Figure 2B shows that DNA synthesis was not modified by low concentration of H2O2 (1 μM), but high concentrations of H2O2 (10–100 μM) did reduce DNA synthesis in a concentration-dependent manner and that decreased DNA synthesis was partially prevented by NAC. The inhibitory effects of H2O2 on proliferation were also evident in floating neurospheres, as shown by reduction in neurosphere number and size in photomicrography. The restorative action of NAC was also evident in floating neurospheres that were treated with H2O2 (Fig. 2C). H2O2 reduced the number (Fig. 2D) and the size (Fig. 2E) of new neurospheres generated 2 days after subculture and total cell numbers per well in a concentration-dependent manner (Fig. 2F). When the neurospheres were pretreated with NAC before exposure to H2O2, however, NSC proliferation was restored, by showing increase in newly formed neurosphere number, size, and total cell numbers per well (Fig. 2G–2I).
![H2O2-treated Atm+/+ neural stem cells have enhanced intracellular ROS levels and show reduced proliferation. (A and B): Atm+/+ neural stem cells (NSCs) were treated with the indicated concentrations of H2O2, either in the absence or presence of 1 mM NAC, and intracellular 2′-7′-dichlorofluorescein diacetate levels and [3H]-thymidine uptake were measured. The mean ± SE of three independent experiments is shown. *, p < .01; ***, p < .001, when H2O2-treated Atm+/+ NSCs were compared with untreated NSCs or when H2O2/NAC-treated Atm+/+ NSCs were compared with H2O2-treated NSCs. (C): Photomicrograph of Atm+/+ neurospheres treated with 50 μM H2O2, either in the absence or presence of NAC. Scale bars = 50 μm. (D–F): Neurospheres were treated with the indicated concentrations of H2O2, and the following were assessed: (D) numbers of newly formed neurospheres, (E) size of newly formed neurospheres, and (F) total cell numbers per well. The mean ± SE of three independent experiments is shown. *, p < .05; **, p < .01; ***, p < .001, when H2O2-treated Atm+/+ NSCs were compared with untreated Atm+/+ NSCs. (G–I): Atm+/+ NSCs were treated with 50 μM H2O2, either in the absence or presence of NAC, and the following were assessed: (G) numbers of newly formed neurospheres, (H) size of newly formed neurospheres, and (I) total cell numbers per well. The mean ± SE of three independent experiments is shown. **, p < .01, when H2O2-treated Atm+/+ NSCs were compared with untreated NSCs or when H2O2/NAC-treated Atm+/+ NSCs were compared with H2O2-treated Atm+/+ NSCs. Abbreviations: NAC, N-acetyl-L-cysteine; ROS, reactive oxygen species.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/stmcls/27/8/10.1002_stem.125/3/m_stmcls_27_8_1987_nfig002.jpeg?Expires=1716312896&Signature=YyihY1uauxuXI7Wuj6q4J9Co6B3SaM0IEXvCennWGFR-0imKHLD~kzeaAz1~AtE5XpXAOBzHNtMe4v-wkeQnLVIJhKIGiK67ox7XuUyz-q4CSfEmmWXteRHCWTCXRzvH-XnU~REaE3aOmKo5Iiv90p1T9ZhPmUMLERVr5JfoAF27V-IzVxRSgyINCfLpvbC9FYBNAvf1SwFKMq7Db~IDekEHcQiLJ-lAp9aI-aHMYtl7SaEmYwYGSnJlRw2yU5OQ~xFSJOUk6RdWAr17s-MfH7ICdGHBJACRJme5pvdJhjandG2wmWrRgpuixQaW34-LTYN-ELvPx55um07inLl2Xg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
H2O2-treated Atm+/+ neural stem cells have enhanced intracellular ROS levels and show reduced proliferation. (A and B): Atm+/+ neural stem cells (NSCs) were treated with the indicated concentrations of H2O2, either in the absence or presence of 1 mM NAC, and intracellular 2′-7′-dichlorofluorescein diacetate levels and [3H]-thymidine uptake were measured. The mean ± SE of three independent experiments is shown. *, p < .01; ***, p < .001, when H2O2-treated Atm+/+ NSCs were compared with untreated NSCs or when H2O2/NAC-treated Atm+/+ NSCs were compared with H2O2-treated NSCs. (C): Photomicrograph of Atm+/+ neurospheres treated with 50 μM H2O2, either in the absence or presence of NAC. Scale bars = 50 μm. (D–F): Neurospheres were treated with the indicated concentrations of H2O2, and the following were assessed: (D) numbers of newly formed neurospheres, (E) size of newly formed neurospheres, and (F) total cell numbers per well. The mean ± SE of three independent experiments is shown. *, p < .05; **, p < .01; ***, p < .001, when H2O2-treated Atm+/+ NSCs were compared with untreated Atm+/+ NSCs. (G–I): Atm+/+ NSCs were treated with 50 μM H2O2, either in the absence or presence of NAC, and the following were assessed: (G) numbers of newly formed neurospheres, (H) size of newly formed neurospheres, and (I) total cell numbers per well. The mean ± SE of three independent experiments is shown. **, p < .01, when H2O2-treated Atm+/+ NSCs were compared with untreated NSCs or when H2O2/NAC-treated Atm+/+ NSCs were compared with H2O2-treated Atm+/+ NSCs. Abbreviations: NAC, N-acetyl-L-cysteine; ROS, reactive oxygen species.
H2O2 Activates Akt, Erk1/2, and p38 MAPK in Atm+/+ Neural Stem Cells
We investigated the effects of H2O2 on several known characteristics of NSCs associated with self-renewal and proliferation. Akt and Erk1/2 have both been implicated in survival of NSCs [14–17], whereas p38 MAPK inhibits NSC proliferation and is involved in NPC fate decisions during neural development [18, 19]. We therefore analyzed the phosphorylation states of Akt, Erk1/2, and p38 MAPK, after H2O2 treatment of Atm+/+ neurospheres, to evaluate its effects on the activity of the PI3K and MAPK signaling pathways, respectively. Figure 3A and 3B shows that H2O2 activated all three kinases in Atm+/+ neurospheres, showing the highest elevation in 1 hour for phospho-Akt and phospho-p38 and in 4 hours for phospho-Erk1/2 after treatment. Interestingly, neurospheres showed constitutive activation of Akt and Erk1/2, but not p38 MAPK, activation, before their exposure to H2O2. However, H2O2 treatment activated Akt, Erk1/2, and p38. Activation of all three kinases was prevented if H2O2 treatment was preceded by NAC treatment, indicating that phosphorylation of these three kinases is ROS dependent (Fig. 3C).
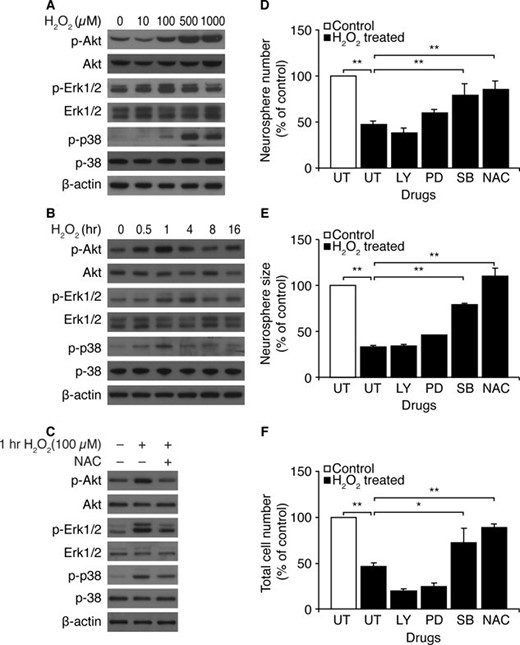
H2O2 activates Akt, Erk1/2, and p38 mitogen-activated protein kinase in Atm+/+ neural stem cells. (A): At 1 hour after exposure to different concentrations of H2O2, levels of pAkt, Akt, pErk1/2, Erk1/2, pp38, and p38 were determined by Western blotting analysis. (B): After exposure to 100 μM H2O2, levels of pAkt, Akt, pErk1/2, Erk1/2, pp38, and p38 were measured by Western blotting analysis at the indicated times. (C): Neurospheres were treated with 50 μM H2O2, either in the absence or presence of NAC, and levels of pAkt, Akt, pErk1/2, Erk1/2, pp38, and p38 were measured at 1 hour after exposure to H2O2. (D–F): NSCs were pretreated with LY294002, PD98059, SB203580, or NAC. Proliferation of NSCs was determined in the presence of 50 μM H2O2 by assessing (D) numbers of newly formed neurospheres, (E) size of newly formed neurospheres, and (F) total cell numbers per well. The mean ± SE of three independent experiments is shown. *, p < .05; **, p < .01, when H2O2-treated Atm+/+ NSCs were compared with untreated Atm+/+ NSCs or when H2O2/drug-treated Atm+/+ NSCs were compared with H2O2-treated Atm+/+ NSCs. Abbreviations: LY, LY294002; NAC, N-acetyl-L-cysteine; PD, PD98059; SB, SB203580; UT, untreated.
To investigate the roles of Akt and MAPK signaling pathways in NSC proliferation, dissociated NSCs were pretreated with the PI3K inhibitor LY294002, the MEK-specific MAPK inhibitor PD98059, or the p38 MAPK inhibitor SB203580 before H2O2 treatment. The numbers and sizes of neurospheres, in addition to total cell number, were compared 48 hours later. Ten micromolar LY294001, 50 μM PD98059, and 10 μM SB203580 inactivated Akt, Erk1/2, and p38 in the neurospheres, respectively (supporting information Fig. 2). Of the three inhibitors, only SB203580 improved newly formed neurosphere numbers, sizes, and total cell numbers to levels similar to those of untreated controls. In contrast, LY294002 reduced newly formed neurosphere numbers and total cell numbers per well but had no effect on neurosphere size. PD98059 slightly improved numbers and sizes of newly formed neurospheres, although it slightly reduced total cell number (Fig. 3D–3F). Interestingly, even though H2O2 treatment affects both Akt and Erk1/2 in neurospheres (Fig. 3A and 3B), proliferation analysis of cells treated with kinase-specific inhibitors indicates that neither pathway is involved in ROS-mediated defects of NSC proliferation. It is possible that short-term ROS-induced activation of Akt and Erk1/2 might occur and might play a protective role against ROS. Nevertheless, our results are consistent with previous reports by others showing that Akt is essential for neurosphere formation and that the Erk1/2 pathway contributes to neurosphere growth [14–17]. However, our results also show that inhibition of p38 MAPK rescued neurosphere numbers and sizes, as well as total cell numbers, indicating that p38 MAPK may contribute to ROS-induced proliferation defects in neurospheres.
H2O2 Augments Expression of the Cyclin-Dependent Kinase Inhibitors p21Cip1 and p27Kip1 in Atm+/+ Neural Stem Cells
Proliferation induced by the PI3K pathway requires Akt-mediated phosphorylation of the cyclin-dependent kinase (CDK) inhibitor p27Kip1, which is retained in the cytosol. In the absence of mitogens, p27Kip1 enters the nucleus, where it prevents cell cycle progression. For this reason, CDK inhibitors may determine NSC fate. For example, p27Kip1 is expressed in the SVZ of adult mice [24], where it directs the cells toward self-renewal and differentiation, via regulating the length of the G1 phase of the cell cycle in central nervous system progenitors [25, 26]. p16 INK4a also decreases NPCs during aging [20], and elevated ROS levels decreases life span of hematopoietic stem cells (HSCs) [27]. Therefore, we decided to investigate the effects of oxidative stress on levels of CDK inhibitors in NSCs. Figure 4A shows that H2O2 elevated levels of p21cip1 and p27kip1, but not p16Ink4a, in neurospheres. However, levels of cyclin E and cyclin D1 decreased in response to H2O2 (Fig. 4B). These alterations were restored by NAC, indicating that these events were ROS mediated (Fig. 4C). Regulation of NSC proliferation via maintaining the redox balance involves the SOD1 enzyme [13, 28]. We therefore assessed expression of SOD1 in response to H2O2 in neurospheres. When exposed to H2O2, Atm+/+ neurospheres contained reduced levels of SOD1, but normal levels were maintained in cultures pretreated with NAC. This observation further supports our above findings that intracellular ROS levels were elevated in NSCs in response to H2O2 (Fig. 2A), with decreased cell proliferation (Fig. 2D–2F). It is also consistent with our immunostaining studies, which showed that p27Kip1 was highly expressed in neurospheres in response to H2O2 but decreased in the presence of NAC (Fig. 4D). To determine whether inhibition of kinases has functional consequences for their downstream effectors, the effects of inhibitors on the levels of p21cip1 and p27kip1 were assessed. Increases of both CDK inhibitors occurred in response to H2O2, but these enhancements were reduced by SB203580. PD98059 decreased p27kip1, not p21cip1, whereas no reduction of p21cip1 and p27kip1 by LY294002 was observed (Fig. 4E). The data indicate that H2O2 acts through p38 MAPK signaling to upregulate p21cip1 and p27kip1 in neurospheres.
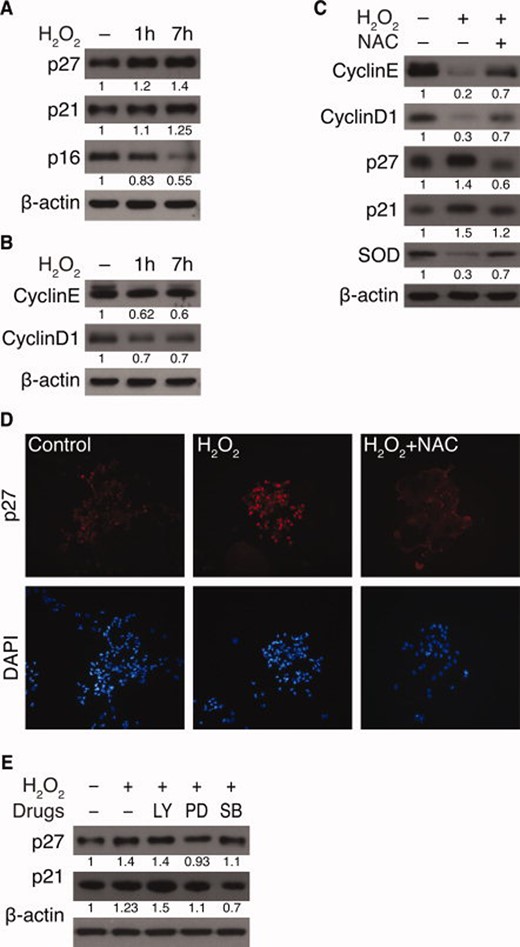
H2O2 augments expression of the cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1 in Atm+/+ neural stem cells. (A and B): Neurospheres were treated with 50 μM H2O2 for indicated times, and the following were measured by Western blotting analysis: (A) levels of p27kip1, p21cip1, and p16Ink4a and (B) levels of cyclin E and cyclin D1. (C): Neurospheres were treated with 50 μM H2O2, either in the absence or presence of NAC, and levels of cyclin E, cyclin D1, p27kip1, p21cip1, and SOD1 were measured. (D): Immunocytochemistry of p27kip1 after treatment of 50 μM H2O2 for 7 hours, either in the absence or presence of NAC. (E): Neurospheres were either left untreated or treated with 50 μM H2O2 in the presence of LY294002, PD98059, or SB203580. Levels of p27kip1 and p21cip1 were measured by Western blotting analysis. Abbreviations: LY, LY294002; NAC, N-acetyl-L-cysteine; PD, PD98059; SB, SB203580; SOD, superoxide dismutase.
In Atm−/− NSCs, p38 MAPK Activation Occurs and CDK Inhibitors Are Upregulated
Atm−/− neurospheres exhibit elevated ROS levels (Fig. 1C). In addition, Akt and Erk1/2 are activated in these cells after H2O2treatment (Fig. 3). In contrast, p38 MAPK activation mediates ROS-induced proliferation defects in neurospheres. We therefore asked whether the absence of ATM alters the activation status of Akt, Erk1/2, and p38 MAPK, by measuring levels of pAkt, pErk1/2, and p38 in Atm−/− neurospheres. Figure 5A shows that p38 MAPK levels are increased, whereas pErk1/2 and pAkt levels are decreased in Atm−/− neurospheres. To assess the effects of these alterations on downstream effectors, the expression levels of CDK inhibitors were determined in Atm−/− neurospheres. Figure 5B shows that basal levels of p21cip1 and p27kip1 are elevated in Atm−/− neurospheres but that p16Ink4a is decreased. This is consistent with protein expression levels of cyclins D1 and E, which were also downregulated in Atm−/− neurospheres. In addition, SOD1 levels were drastically decreased in Atm−/− neurospheres when they were compared with Atm+/+ neurospheres (Fig. 5C). Fluorescence microscopy showed that Atm+/+ and Atm−/− neurospheres both were composed of nestin-positive cells but that Atm−/− neurospheres had reduced expression of Bmi1, which promotes neural cell proliferation, compared with Atm+/+ neurospheres (Fig. 5D). Finally, Atm−/− neurospheres show enhanced activity of p38 MAPK, with concomitant upregulation of p27kip1 (supporting information Fig. 3).
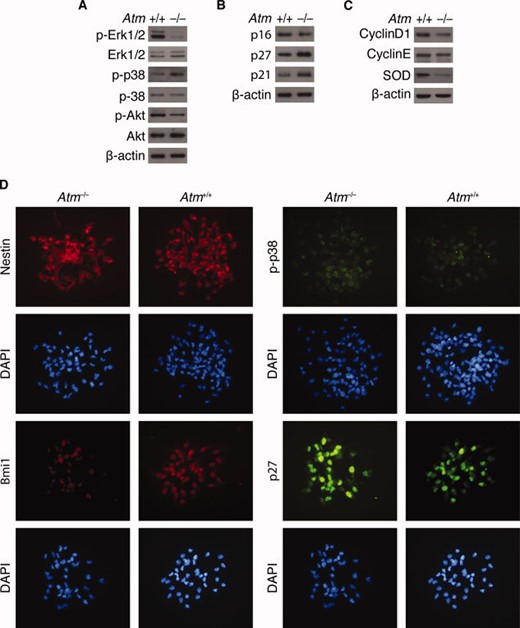
In Atm−/− neural stem cells, p38 mitogen-activated protein kinase activation occurs and cyclin-dependent kinase inhibitors are upregulated. (A–C): Levels of the following proteins in Atm+/+ and Atm−/− neurospheres were measured by Western blotting analysis: (A) pERK1/2, ERK1/2, pp38, p38, pAkt, and Akt; (B) p16Ink4a, p27kip, and p21cip1; and (C) cyclin D1, cyclin E, and SOD1. (D): Immunocytochemistry of nestin, Bmi1, pp38, and p27kip1 in Atm−/− and Atm+/+ neurospheres. Abbreviations: DAPI, 4′, 6-diamidino-2-phenylindole; SOD, superoxide dismutase.
Proliferation of Atm−/− NSCs Is Improved by NAC and by Inhibition of p38 MAPK, via p21cip1/p27Kip1 Downregulation
To determine whether activation of p38 MAPK impairs survival and proliferation of Atm−/− NSCs, Atm−/− NSCs were treated with the p38 MAPK inhibitor SB203580. Figure 6A–6C shows that SB203580 treatment greatly improves newly formed neurosphere number, size, and total cell numbers per well. The rescuing effects of SB203580 on proliferation of Atm−/− NSCs were comparable to those of antioxidant NAC. However, inactivation of Akt by LY294002 reduced neurosphere numbers and total cell numbers per well but had no effect on neurosphere size. Erk1/2 inactivation by PD98059 slightly improved neurosphere formation in Atm−/− NSCs but not neurosphere size or cell division. In accordance with these results, NAC and SB203580 downregulated levels of p21cip1 and p27kip1 in Atm−/− neurospheres (Fig. 6D). It has been shown that PI3K-Akt signaling delays H2O2-induced apoptosis in cultured NSC/NPCs [13]. In addition, PI3K-Akt signaling is essential for maintenance of NSC/NPCs, because it links cell proliferation to inhibition of differentiation by regulating p27kip1 [23]. We therefore asked whether NAC and/or p38 MAPK inhibition activates both Erk1/2 and/or Akt signaling in Atm−/− neurospheres. Figure 6E shows that both NAC treatment and p38 MAPK inhibition by SB203580 promoted activation of Akt and Erk1/2 in Atm−/− neurospheres. The restorative actions of NAC and SB203580 were also evident in floating neurospheres, as shown by increased number and size (Fig. 6F).
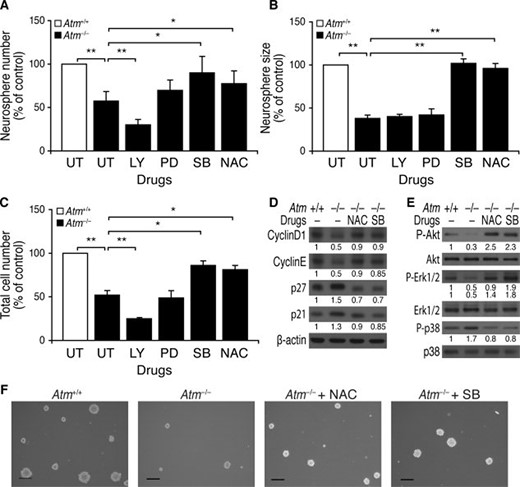
Proliferation of Atm−/− neural stem cells (NSCs) was improved by NAC and by inhibition of p38 mitogen-activated protein kinase via p21cip1/p27Kip1 downregulation. (A–C): Atm+/+ neurospheres were left untreated. Atm−/− neurospheres were either left untreated or treated with LY294002, PD98059, SB203580, or NAC, and the following were assessed: (A) numbers of newly formed neurospheres, (B) size of newly formed neurospheres, and (C) total cell numbers per well. The mean ± SE of three independent experiments is shown. *, p < .05; **, p < .01, when untreated Atm−/− NSCs were compared with untreated Atm+/+ NSCs or when drug-treated Atm−/− NSCs were compared with untreated Atm−/− NSCs. (D–F): Atm+/+ neurospheres were left untreated. Atm−/− neurospheres were either left untreated or treated with NAC or SB203580 for 2 days, and the following were assessed: (D) levels of cyclin D1, cyclin E, p27kip1, and p21cip1; (E) levels of pAkt, Akt, pErk1/2, Erk1/2, pp38, and p38; and (F) photomicrographs of Atm+/+ and Atm−/− neurospheres in the presence of NAC or SB203580. Scale bars = 50 μm. Abbreviations: LY, LY294002; NAC, N-acetyl-L-cysteine; PD, PD98059; SB, SB203580; UT, untreated.
Discussion
In the normal brain, the numbers of stem cells in the SVZ are the result of a tightly controlled balance between self-renewal, differentiation, and death [16]. This means that control of proliferation of the neural stem cells/precursor cells plays a critical role in determining the number of neurons, astrocytes, and oligodendrocytes in the brain. In addition, it is increasingly apparent that stem cell proliferation and maturation require supportive microenvironment including astrocytes. Astrocytes have well-established roles in regulating the microenvironment in the central nervous system, including redox homeostasis. Astrocytes in the hippocampus and SVZ also support stem cell proliferation and maintenance [29–31]. We have recently shown that ATM is required to maintain survival and proliferation of astrocytes by controlling the redox status of these cells [32]. ATM has also been shown by others to play an important role for HSC survival and lifespan [27]. However, despite its importance, the effects of ATM deficiency in NSC survival and proliferation remain unclear.
In this study, we investigated the effects of ATM deficiency on NSC survival, focusing specifically on NSC proliferation. The data presented here showed that defective proliferation of NSC is part of the neurodegenerative phenotype in Atm−/− mice and that oxidative stress contributes to defective NSC proliferation. In these studies, however, we also uncovered an intrinsic defect in Atm−/− NSCs, by showing that they survive poorly in culture and are markedly deficient in proliferation. Human A-T disease shares clinical features with other genetic disorders, such as the Nijmegen breakage syndrome (caused by a hypomorphism in NBS1) [33] and ataxia-telangiectasia-like disorder (caused by a hypomorphism in MRE11A) [34]. Interestingly, Nbn-deficient NSCs show proliferation defects, as do Atm−/− NSCs, but do not show increased apoptosis [35]. In our study, analysis of apoptosis by the TUNEL technique indicated that 7.8% ± 4% (n = 3) of Atm+/+ NSCs and 8.2% ± 5% (n = 3) of Atm−/− NSCs show apoptotic nuclei. In addition, similar levels of cleaved caspase 3 and γ-H2AX were found in Atm+/+ and Atm−/− neurospheres (supporting information Fig. 4). These data indicate that defective proliferation in Atm−/− NSCs, like Nbn-deficient NSCs, may not result from apoptosis of NSCs.
The data presented here has identified signaling pathways that cause defective proliferation in Atm−/− NSCs. The results show that ROS-induced increases in p38 MAPK activity contribute to this phenotype. Previous work by others has shown that activation of the p38 MAPK pathway in the central nervous system induces neural cell apoptosis by causing oxidative stress [18, 19]. Interestingly, another recent study has shown that ROS-mediated p38 MAPK also contributes to defective self-renewal of Atm−/− HSCs [27]. This finding together with our results now suggest that ATM functions in NSC/HSC survival by controlling ROS levels and that this control may be exerted via suppression of the p38 MAPK pathway.
Proliferation and differentiation of NSCs are carefully controlled in normal brains. For our studies, it is significant that p38 MAPK activation is associated with neuronal differentiation [36], whereas this pathway is suppressed in proliferating NSCs. Atm−/− NSC/NPCs show abnormal differentiation [11], but the mechanism for this has been unclear. We propose here that reduction of oxidative stress in Atm−/− NSCs, either with NAC or with a p38 MAPK inhibitor, may promote normal neural differentiation. In this regard, we have previously shown that neurons and astrocytes are both affected in Atm−/− mice [32, 37]. Both defects may be results of decreased survival and impaired proliferation that we observed in Atm−/− NSCs.
We also show here that Erk1/2 and Akt are transiently activated by H2O2 in Atm+/+ neurospheres, whereas activation levels of both kinases are constitutively downregulated in Atm−/− neurospheres. Furthermore, Erk1/2 inhibition does not rescue proliferation of Atm−/− NSCs, and Akt inhibition exacerbates defects in proliferation of Atm−/− NSCs. These findings are consistent with work by others showing that both Erk1/2 and Akt signaling play pivotal roles in NSC/NPC proliferation [38, 39]. The data also show that Erk1/2 is required both for the proliferation of NSC and for the maintenance of NPC multipotency, because it suppresses the commitment of these cells to a glial lineage [17].
Conclusion
We show here that NSCs from Atm−/− mice have intrinsic impairments in their survival and proliferation. We also show that oxidative stress-mediated p38 MAPK signaling exacerbates these defects. We conclude that ATM is an essential participant in controlling normal proliferation of NSCs. Based on this conclusion, we believe that controlling oxidative stress might be therapeutically useful in the treatment of A-T neurodegeneration. Another possible therapeutic tool for A-T involves transplantation of normal NSCs to A-T patients, to generate new neurons or to replace degenerated cells. With a better understanding of the mechanisms by which ATM regulates endogenous NSC functions, we should have improved insights into the possible application of stem cell therapies in A-T neurodegeneration.
Acknowledgements
We thank Dr. D. Tang and Dr. V. L. Scofield for critically reviewing this manuscript and Dr. C. Rothblum-Oviatt for her comments. This project was supported by a grant from the Odyssey Program of the M.D. Anderson Cancer Center to J.S.K. and by funds from the Longevity Foundation (Formally, A-T foundation) in Austin, TX, and the A-T Children's Project in Deerfield Beach, FL.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
Author notes
Author contributions: J.K.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; P.K.Y.W.: conception and design, provision of study materials, financial support.
First published online in STEM CELLS EXPRESS May 14, 2009.
Telephone: 1-512-237-9456; Fax: 1-512-237-2444
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}